



药物相互作用研究
对于在研药物来说,评估其药物相互作用是评价药物安全性的重要一环,EMA,PMDA, FDA 和 NMPA均发布了对应的指导原则来具体阐述需要开展的研究。DDI评估通常从体外试验开始,确定可能影响药物处置的因素以阐明潜在的DDI机制,其主要内容包括评估代谢酶和转运体介导的相互作用。

-
平台介绍
-
代谢相关的药物相互作用研究
-
转运体介导的药物相互作用
-
案例分享
-
经验
-
常见问答
-
相关资源
-
相关服务



平台介绍
DDI评估通常从体外试验开始,确定可能影响药物处置的因素以阐明潜在的 DDI机制,体外试验的数据可以有效的支持IND和NDA阶段的申报工作。为了保证客户实验数据的准确性及可靠性,体外DDI的标准实验体系均经过了验证,并用指导原则推荐的阳性对照化合物进行质控。
药明康德药物代谢与动力学部的DDI平台凭借完善的药物-药物相互作用性质研究平台和上千个申报项目的经验,可以满足各个监管机构的申报要求。
Learn More


代谢相关的药物相互作用研究
代谢酶介导的DDI评估通常从体外试验开始,确定可能影响药物处置的因素以阐明潜在的DDI机制,并获得用于进一步研究的动力学参数,其主要内容包括:确定药物的主要消除途径;评估相关代谢酶对药物处置的贡献;考察药物对代谢酶的影响。药明康德体外代谢酶介导的药物相互作用平台提供广泛的服务,包括:cytochrome P450 抑制, CYP450 时间依赖性抑制, CYP450 诱导(酶活和/或基因水平测试,加上 RIS 方法评估), UGT 诱导和抑制研究,也包括一些其他代谢酶的相应的研究。作为深入的机制研究,我们还提供Ki和Kinact/KI的研究以明确可逆抑制和时间依赖性抑制的机理。在代谢酶表型鉴定方面,我们有对应的CYP450酶和UGT酶的完整的表型鉴定服务以及其他的代谢酶的表型鉴定服务。
了解更多信息
下面列出了更详细的分析类型:
-
Cytochrome P450 (CYP) 酶抑制
CYP1A2、CYP2A6、CYP2B6、 CYP2C8、CYP2C9、CYP2C19、CYP2D6、CYP2E1和CYP3A
直接抑制
用单一/5合1/7合1的探针底物进行IC50测定,用单一的探针底物进行Ki 或Ki,u测定
时间依赖性抑制
用单一的探针底物进行IC50 shift、AUC shift、Kinact/KI测定


-
Cytochrome P450 (CYP) 酶诱导
受体激活检测
人肝细胞体系(CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19和CYP3A4)酶活和/或基因水平测试
EC50 和Emax 测定
RIS 评估
-
酶反应表型鉴定
Cytochrome P450 (CYP)酶表型鉴定
UDP-Glucuronosyl transferases 酶表型鉴定
AO, CES,MAO, FMO, XO和ADH/ALDH 酶反应表型鉴定

-
UGT酶抑制或诱导研究
UGT 抑制
用重组人UGT酶测定IC50 ,用人肝微粒体测定IC50
UGT 诱导
mRNA 评估

转运体介导的药物相互作用
体外转运体研究平台提供多种模型进行特定转运体底物或者抑制剂的评估,1)Caco-2细胞和2)MDR1-MDCK I和MDR1-MDCK II细胞,用于开展P-gp相关药物相互作用研究。其中,MDR1-MDCK I细胞可以更准确地预测体内血脑屏障的P-gp底物,主要应用于中枢神经相关的药物研发。3)稳定转染人源OATP1B1、OATP1B3、OATP2B1、OAT1、OAT3、OCT1、OCT2、MATE1、MATE2-K、PEPT1、PEPT2和NTCP基因的HEK293细胞,用于开展SLC转运体的药物相互作用研究。此外,我们还提供表达ABC转运体P-gp、BCRP、BSEP和MRP1/2/3/4的囊泡模型,开展相关研究工作,并建立了利用原代肝细胞综合评估肝脏的摄取能力及摄取转运体介导的药物相互作用模型。
关于转运体介导的药物相互作用服务详情,请见渗透性与转运体服务研究页面。
案例分享
-
Cytochrome P450 抑制实验
药明康德DMPK的CYP酶抑制平台目前有三种独立的体系开展,依据客户的不同目的开展。 所有的实验体系均采用商品化标准品进行详尽的验证工作,如下展示了不同体系测得的IC50之间的相关性,图1展示了7合1 混合探针底物与单一探针底物两种方法测得的IC50之间的相关性强(相关系数的平方值(R2)为0.995)。我们也将内部体系产生的数据与文献数据进行了对比,如图2所示,5合1 混合探针底物体系内部测得的IC50与文献报道数据也相关性较强。
Learn More
-

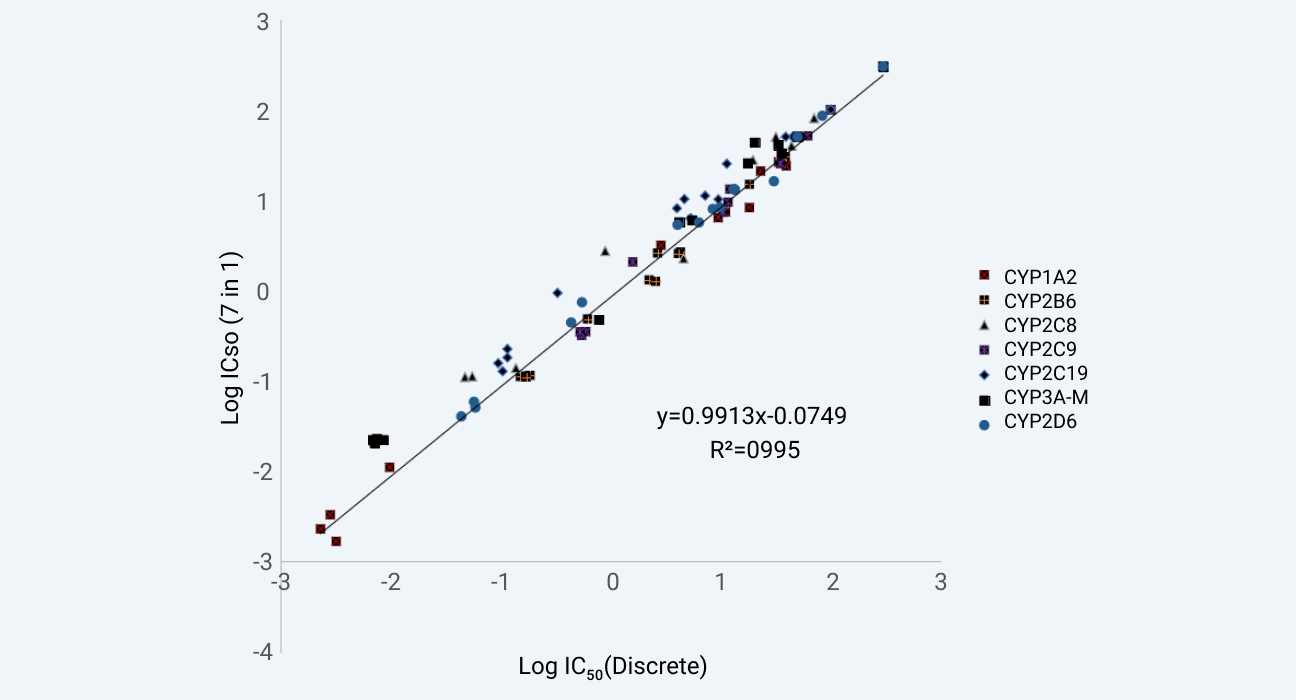
药明康德DMPK自研 7合1底物与单一底物评估CYP酶抑制能力的相关性研究
图1
-
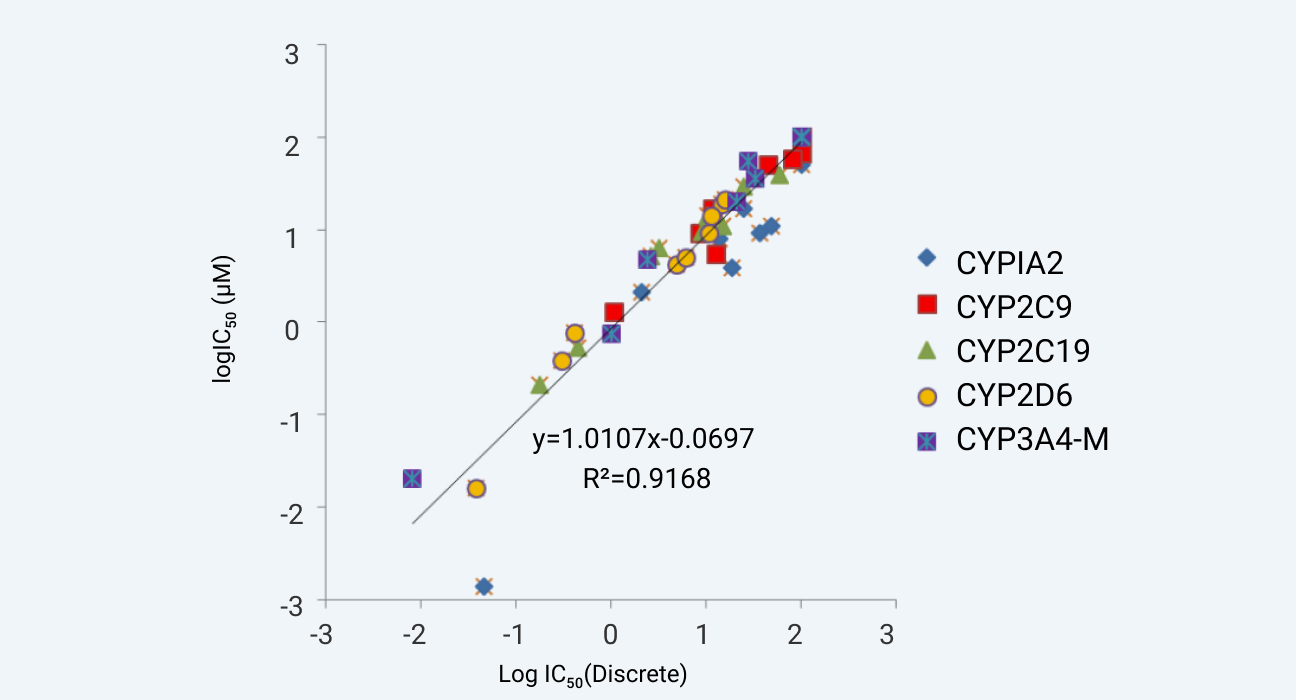
药明康德DMPK自研 5合1底物评估CYP酶抑制能力与文献报道值的相关性研究
图2
-
经验
-
15+
年


-
1,500+
IND申报项目

-
3,000+
筛选项目每年

常见问答
-

什么是药物相互作用?


药物相互作用是指联合用药时,一种药物的作用受其他药物的影响而发生改变,导致药物作用增强、减弱或出现新的作用。
而广义的药物相互作用,不仅包括药物和药物之间的相互作用,还包括药物和食物及饮料之间的相互作用,甚至还包括药物和疾病治疗状态下的相互作用。
-
CYP酶诱导剂判断标准:mRNA水平 or 酶活性水平?
1.根据指导原则的变化历程来看,酶活性自始至终都是备受关注且广泛使用的指标。
2.从中心法则的角度来说,mRNA的水平变化会早于酶活性的变化,因而早期的筛选实验可以仅采用mRNA水平评估。
3.单一的指标容易受不同的影响因素干扰,酶活性指标容易受化合物本身是抑制剂干扰,而mRNA指标容易受化合物存在基因下调可能性的影响,因而申报阶段需要同时考察两个指标。
-
药物相互作用(DDI)的类型有哪些?
按照发生的原理可以分为药代动力学相关和/或药效学相关的药物相互作用,其中药代动力学相关的药物相互作用在药物开发过程中更受重视,根据作用机制可以分为以下两个大类:
1. 代谢酶介导:细胞色素P450酶系抑制或诱导,或二相酶UGT酶的抑制等(如:葡萄柚汁抑制CYP3A4酶活性,从而使得多达85种药物被发现与葡萄柚同服会引发不良反应)
2. 转运体介导:ABC类转运体或SLC类转运体的抑制,从而影响药物吸收/排泄其他重要考量:除了对代谢酶或转运体有抑制或诱导外,同时还要考虑药物被代谢酶代谢或者是转运体的底物的可能性。
-
什么阶段需要开展药物相互作用研究?
根据《ICH M12:药物相互作用研究》(NMPA 2024),DDI研究应贯穿药物开发全过程,主要分为3个阶段:
1) 临床前识别,即体外实验
1. 体外研究是确定药物作为DDI促变药或受变药风险的首要步骤,包括对药物代谢酶的抑制或诱导作用以及通过体外酶表型实验识别在研药物的主要代谢清除途径。
2. 体外研究应使用转运蛋白活性经探针底物和抑制剂验证的试验系统来考察在研药物是否为转运体的底物;此外,应采用同样的试验系统开展研究评价在研药物是否为P-gp、BCRP、OATP1B1、OATP1B3、OAT1、OAT3、OCT2、MATE1和MATE2-K的抑制剂。必要时,可考虑评价药物对其他转运体的抑制潜力。如果联用的药物是其他转运体的底物,则可以考虑评价这些转运体。
2) 临床阶段识别,即临床评价
有多种研究类型可用于评价临床DDI发生的可能性及其严重程度。在进行DDI研究时,应根据研究的具体目的,选择适当的研究类型。
在某些情况下,可基于体外试验结果使用模型预测的方法(静态机制模型或PBPK模型)预测潜在的临床DDI,而无需进行额外的临床DDI研究。
3) 上市后监测
DDI是临床合理用药和上市后药物警戒中最重要的问题之一。上市后监测药物相互作用可以了解新药与其他药物合用时是否会产生预期之外的效果变化或者是增加不良反应的风险。
相关资源


-

能否根据直接抑制(DDIM)结果判定时间依赖性抑制(TDI)风险?
 文章2025-12-19查看更多
文章2025-12-19查看更多
-
解密血浆蛋白结合(PPB)在药物相互作用中的关键角色
文章2025-08-15查看更多 -
CYP4酶的体外抑制研究策略及抑制剂筛选平台
学术海报2024-12-03查看更多 -
UGT1A1活性种属差异和不同种属抑制剂筛选体外研究
学术海报2024-12-03查看更多 -
肝脏转运体在药物性肝损伤(DILI)研究中的作用简介
文章2024-08-09查看更多 -
ABC转运体体外研究模型的应用及策略
文章2024-07-18查看更多 -
“罕见”CYP酶体系如何完善酶表型鉴定?
文章2024-05-06查看更多 -
CYP酶诱导剂判断标准:mRNA水平 or 酶活性水平?
文章2024-04-18查看更多 -
在CYP酶诱导实验中,如何获得更准确的EC50和Emax值?
文章2024-03-07查看更多 -
体外评估时间依赖性抑制TDI的新方法:AUC Shift法
文章2023-11-30查看更多 -
PROTAC该如何评估外排转运体介导的药物相互作用
文章2023-07-07查看更多 -
评估代谢酶及转运体介导的药物相互作用
网络研讨会2023-06-14查看更多 -
解读丨NMPA,FDA药物相互作用研究技术指导原则(一)──代谢酶介导的药物相互作用评估策略
文章2023-05-17查看更多 -
解读丨NMPA,FDA药物相互作用研究技术指导原则(二)──转运体介导的药物相互作用体外评估策略
文章2023-05-17查看更多


















加入订阅
获取药物代谢与药代动力学最新专业内容和信息
