





2024年,FDA批准了50款小分子和生物大分子新药,这些新药涵盖了多种适应症和分子类型[1]。本文将挑选12个具有代表性的药物,详细分析它们的药物代谢和药代动力学(DMPK)及药物相互作用特征。这些药物包括首款治疗非酒精性脂肪性肝炎(NASH)/代谢功能障碍相关脂肪性肝炎(MASH)的药物Rezdiffra、突破性抗肿瘤药物Lazcluze、创新精神分裂症治疗药物Cobenfy、阿尔茨海默病治疗药物Kisunla、囊性纤维化治疗药物Alyftrek、中枢神经系统肿瘤药物Voranigo、心血管疾病治疗药物Tryvio、骨髓增生异常综合征创新疗法Rytelo、靶向肝病的寡核苷酸药物Tryngolza、新型抗肿瘤单克隆抗体Tevimbra、小细胞肺癌治疗的双特异性抗体药物Imdelltra和肺动脉高压疾病的突破性药物Winrevair。通过这篇文章,我们希望为读者提供一个清晰、详细的2024年FDA获批新药的药代动力学及药物相互作用概述,并展示这些新药在不同适应症和分子类型中的应用情况。
2024年FDA获批新药概况
2024年FDA药物评价与研究中心(Center for Drug Evaluation and Research, CDER)批准了50种新药上市(图1),比2023年的55种略少,但十年滚动平均值达到了46.5种,创下新高。其中,56%的新药获得优先审评,52%获得孤儿药认定,36%获得突破性疗法认定,14%基于替代终点获准加速审评。从分子类型看,约五分之三的新药为小分子药物(60%),小分子以抗肿瘤的药物为主(23%)[2],其余为新分子实体类药物。
-
生物制剂和抗体:批准的50种新药中,16种为生物制剂(包括13种单克隆抗体和双克隆抗体,其中一款为来自百济神州研发的替雷利珠单抗),占比超过25%[3]。
-
聚乙二醇化药物和氘代药物:批准了2种氘代药物和2种含大分子PEG的肽类药物,再次表明这些策略在提高代谢稳定性和延长药物半衰期方面的出色表现[3]。
-
含氟药物和N-芳香杂环化合物:30种小分子药物中,约三分之二含有氟或N-芳香杂环化合物[3]。
-
组合药物:批准了5种含多个活性成分的组合药物[3]。
-
天然产物药物:一些新药开发基于已知的天然产物结构(如头孢菌素和青霉素)[3]。
-
影像诊断药物:3种药物用于影像诊断[3]。
-
多样性:药物涵盖多种治疗领域(表1,图2),如肿瘤、皮肤、血液、中枢神经系统疾病和肝病等[2]。

图1. 按照分子类型汇总的获批药物数量
|
疾病领域 |
总数 |
小分子 |
抗体 |
其他 |
|
抗肿瘤 |
14 |
7 |
6 |
1 (细胞因子融合蛋白) |
|
中枢神经药物 |
2 |
1 |
1 |
0 |
|
皮肤疾病 |
5 |
2 |
2 |
1 (蛋白质毒素) |
|
血液病 |
5 |
0 |
4 |
1(寡核苷酸) |
|
肾病 |
1 |
1 |
0 |
0 |
|
心血管疾病 |
4 |
3 |
0 |
1 (反义寡核苷酸) |
|
肝病 |
3 |
3 |
0 |
0 |
|
呼吸系统疾病 |
2 |
2 |
0 |
0 |
|
内分泌疾病 |
3 |
2 |
0 |
1 (多肽) |
|
感染性疾病 |
3 |
3 |
0 |
0 |
|
其他类型 |
8 |
6 |
0 |
2 |
表1. 按照疾病领域汇总的获批药物数量

图2. 按照分子类型及疾病领域汇总的获批药物情况
总体来看,2024年FDA的药物批准展现了生物制剂、TIDES药物、氟和N-芳香杂环化合物在药物开发中的重要性,以及组合药物和影像诊断药物的持续发展。
典型获批药物的药代动力学及药物相互作用特征
01 Lazcluze™ (lazertinib)
适应症:非小细胞肺癌
作用机制:表皮生长因子受体(EGFR)的激酶抑制剂,与Rybrevant(amivantamab)联合治疗具有EGFR突变的非小细胞肺癌
分子类型:小分子药物
获批日期:2024年8月19日
开发公司:强生
给药途径:口服
给药制剂:片剂
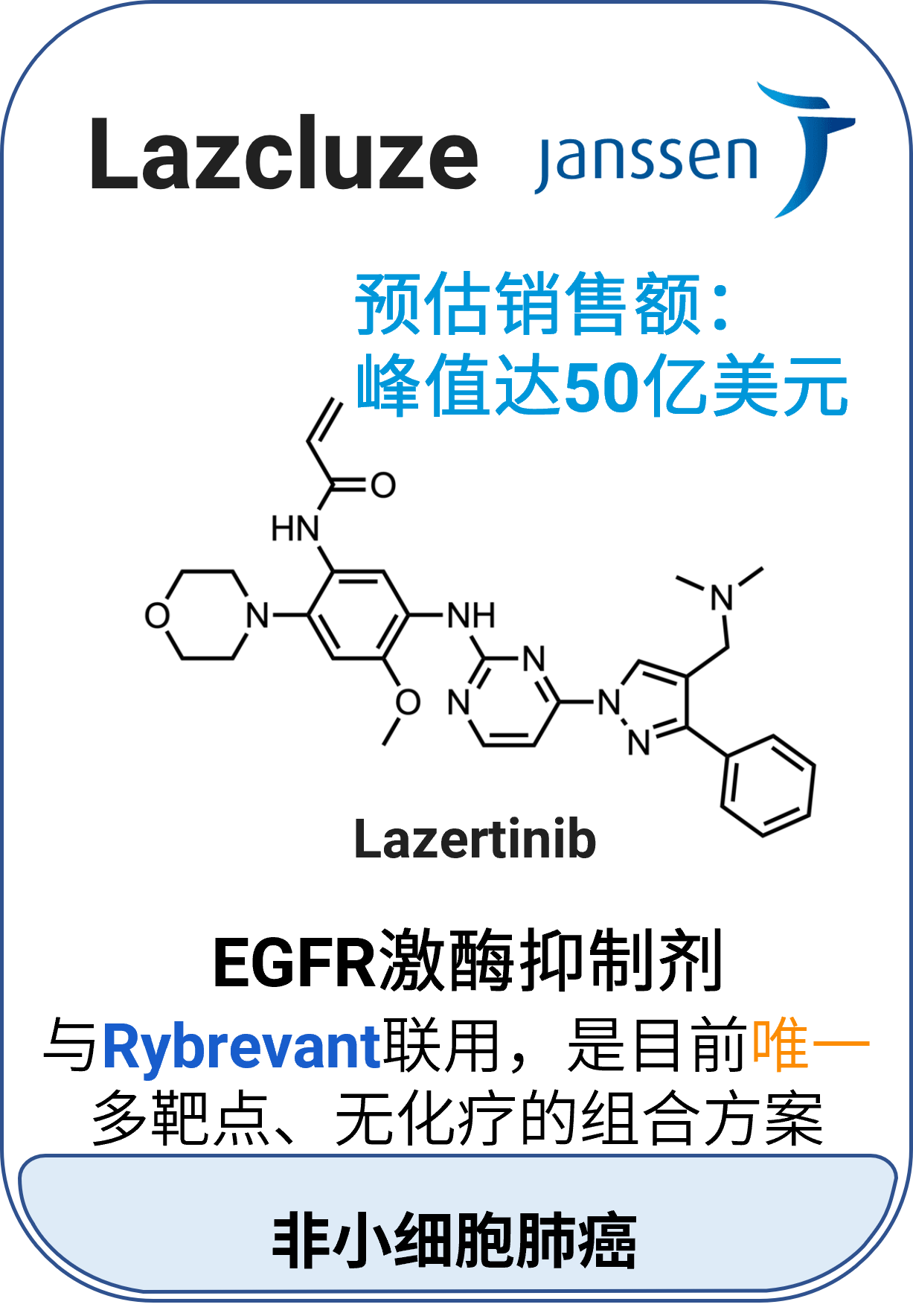
药代动力学及药物相互作用特征:
吸收
Lazertinib单次给药和每日一次给药后,从20 mg至320 mg(0.08至1.3倍的批准推荐剂量)的Cmax和AUC 与剂量成比例增加,血浆暴露量于第15天达到稳态,AUC蓄积约2倍。Tmax: 2-4小时。
分布
分布容积: 2680 L (51% CV);血浆蛋白结合率: 99.2%。
代谢
半衰期: 3.7天 (56% CV);清除率: 36.4 L/h (47% CV)。主要通过谷胱甘肽结合和CYP3A4代谢。
排泄
尿液: 4%(<0.2%原药);粪便: 86%(<5%原药)。
药物相互作用体外研究表明,lazertinib抑制CYP3A4、UGT1A1、BCRP和OCT1。
与强CYP3A4诱导剂(如rifampin)联用时,lazertinib Cmax减少72%,AUC减少83%;与强CYP3A4抑制剂(如itraconazole)联用时,lazertinib Cmax增加1.2倍,AUC增加1.5倍;与CYP3A4底物(如midazolam)联用时,midazolam Cmax增加1.4倍,AUC增加1.5倍;与BCRP底物(如rosuvastatin)联用时,rosuvastatin Cmax增加2.2倍,AUC增加2倍。
02 Cobenfy™ (xanomeline和trospium chloride)
适应症:成人精神分裂症
作用机制:Xanomeline是中枢神经系统M1/M4毒蕈碱激动剂;Trospium chloride是毒蕈碱拮抗剂,主要在外周组织中拮抗毒蕈碱受体
分子类型:小分子组合药物
获批日期:2024年9月26日
开发公司:百时美施贵宝
给药途径:口服
给药制剂:胶囊
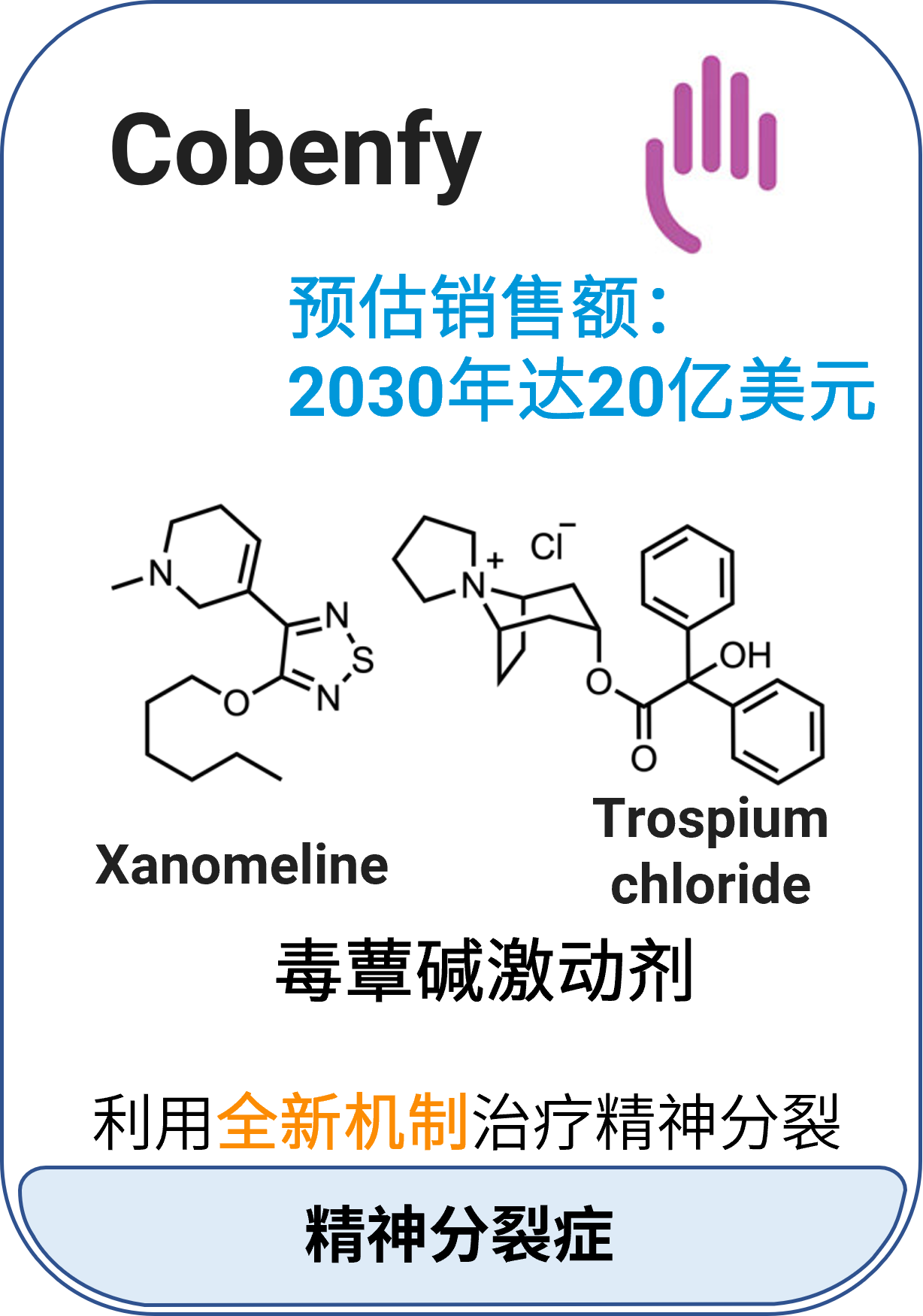
药代动力学及药物相互作用特征:
吸收
在Cobenfy(xanomeline和trospium chloride)剂量从100 mg/20 mg每天两次增加到125 mg/30 mg每天两次时,xanomeline的暴露量增加了50%,trospium的暴露量呈剂量比例增加。
-
Xanomeline:Tmax: 2小时;食物影响:与空腹状态相比,高脂餐后给药的AUC提高30%,Cmax未改变;低脂餐后给药的AUC和Cmax未改变。
-
Trospium chloride:Tmax: 1小时;食物影响:与空腹状态相比,高脂餐和低脂餐后给药的AUC和Cmax都减少85-90%和70-75%。
分布
-
Xanomeline:中央室分布容积: 10800 L;血浆蛋白结合率: 约95%。
-
Trospium chloride:中央室分布容积: 531 L;血浆蛋白结合率: 约80%。
代谢
-
Xanomeline:半衰期: 5小时;总清除率: 1950 L/h;肾清除率: 0.085 L/h;由CYP2D6、CYP2B6、CYP1A2、CYP2C9和CYP2C19、黄素单加氧酶(FMO1和FMO3)代谢。
-
Trospium chloride:半衰期: 6小时;总清除率: 796 L/h;肾清除率: 21 L/h;由酯水解和葡萄糖醛酸结合代谢。
排泄
-
Xanomeline:尿液: 78%(<0.01%原药);粪便: 12%。
-
Trospium chloride:尿液: 85-90%原药。
药物相互作用
体外研究表明,xanomeline不会全身抑制CYP3A4和P-gp,但给药后可能会短暂抑制肠道局部的CYP3A4和P-gp。
当与通过主动肾小管分泌消除的药物、强CYP2D6抑制剂、CYP3A4或P-gp的敏感底物、抗毒蕈碱药物合用时,需要监测相关的不良反应的频率和/或严重程度是否增加。
03 Alyftrek™ (vanzacaftor、tezacaftor和deutivacaftor)
适应症:囊性纤维化
作用机制:囊性纤维化跨膜传导调节因子(CFTR)调节剂,三种成分联合作用增加细胞表面CFTR活性
分子类型:小分子组合药物
获批日期:2024年12月20日
开发公司:Vertex Pharmaceuticals
给药途径:口服
给药制剂:片剂
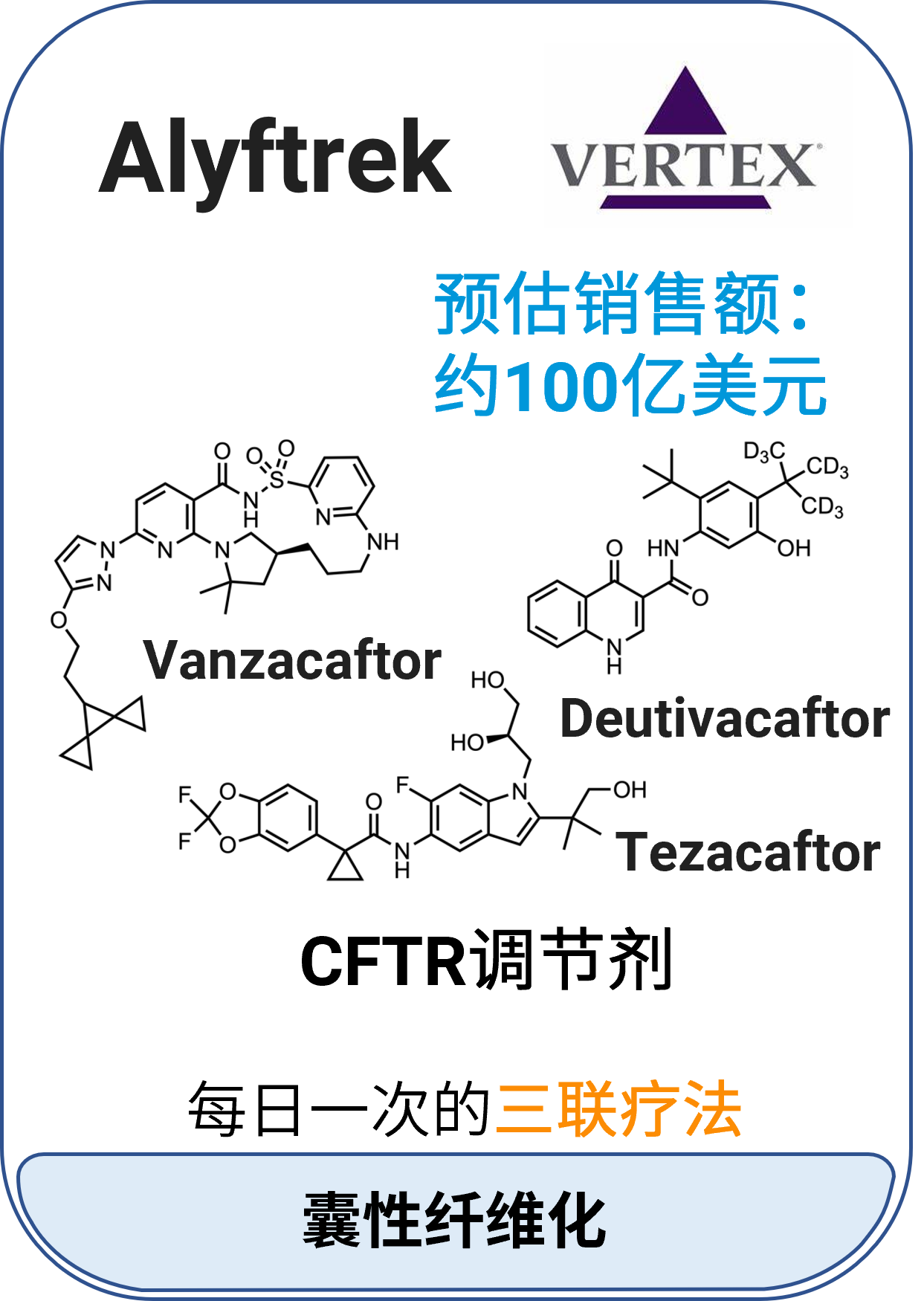
药代动力学及药物相互作用特征:
吸收
在12岁及以上患者的给药剂量(vanzacaftor 20 mg,tezacaftor 100 mg,deutivacaftor 250 mg,每日一次)下,暴露量如下:
-
Vanzacaftor:Cmax,ss: 0.812 μg/mL;AUC0-24h,ss: 18.6 μg·h/mL;Tmax: 7.80小时(3.70至11.9小时)。食物影响:低脂餐下AUC增加4倍,高脂餐下AUC增加6倍。
-
Tezacaftor:Cmax,ss: 6.77 μg/mL;AUC0-24h,ss: 89.5 μg·h/mL;Tmax: 1.60小时(1.40至1.70小时)。食物影响:无显著变化。
-
Deutivacaftor:Cmax,ss: 2.33 μg/mL;AUC0-24h,ss: 39.0 μg·h/mL;Tmax: 3.7小时(2.7至11.4小时)。食物影响:低脂餐下AUC增加3倍,高脂餐下AUC增加4倍。
分布
-
Vanzacaftor:分布容积: 121 L;血浆蛋白结合率: >99%。
-
Tezacaftor:分布容积: 73.1 L;血浆蛋白结合率: 约99%。
-
Deutivacaftor:分布容积: 159 L;血浆蛋白结合率: >99%。
代谢
-
Vanzacaftor:半衰期: 92.8小时;清除率: 1.34 L/h;主要通过CYP3A4/5代谢。
-
Tezacaftor:半衰期: 22.5小时;清除率: 1.22 L/h;主要通过CYP3A4/5代谢。
-
Deutivacaftor:半衰期: 19.2小时;清除率: 7.29 L/h;主要通过CYP3A4/5代谢。
排泄
-
Vanzacaftor:粪便: 91.6%(主要为代谢物);尿液: 0.50%。
-
Tezacaftor:粪便: 72%(原药或M2-TEZ);尿液: 13.7%。
-
Deutivacaftor:未提供。
药物相互作用
体外研究表明:
-
Vanzacaftor:是CYP3A底物,不抑制CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6,不诱导CYP3A4;不抑制OATP1B1和OATP1B3,但抑制BCRP和P-gp。
-
Tezacaftor:是CYP3A底物,不抑制CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6,不诱导CYP3A4;是P-gp、BCRP 和OATP1B1底物,不抑制OATP1B1和OATP1B3,但抑制P-gp。
-
Deutivacaftor:是CYP3A底物,抑制CYP2C8、CYP2C9和CYP3A4,不诱导CYP3A4;是P-gp底物,不抑制OATP1B1和OATP1B3,但抑制P-gp和BCRP。
与强效和中效CYP3A诱导剂合并使用会降低vanzacaftor、tezacaftor 和deutivacaftor的暴露量,可能会降低Alyftrek的疗效。因此,不建议合并使用。与强效或中效CYP3A抑制剂同时使用会增加vanzacaftor、tezacaftor和deutivacaftor的暴露量,可能会增加Alyftrek相关不良反应的风险。因此伴随使用时降低Alyftrek剂量。
04 Rezdiffra™ (resmetirom)
适应症:非肝硬化性非酒精性脂肪性肝炎(NASH)/代谢功能障碍相关脂肪性肝炎(MASH)
作用机制:甲状腺激素受体-β(THR-β)激动剂
分子类型:小分子药物
获批日期:2024年3月14日
开发公司:Madrigal Pharmaceuticals
给药途径:口服
给药制剂:片剂
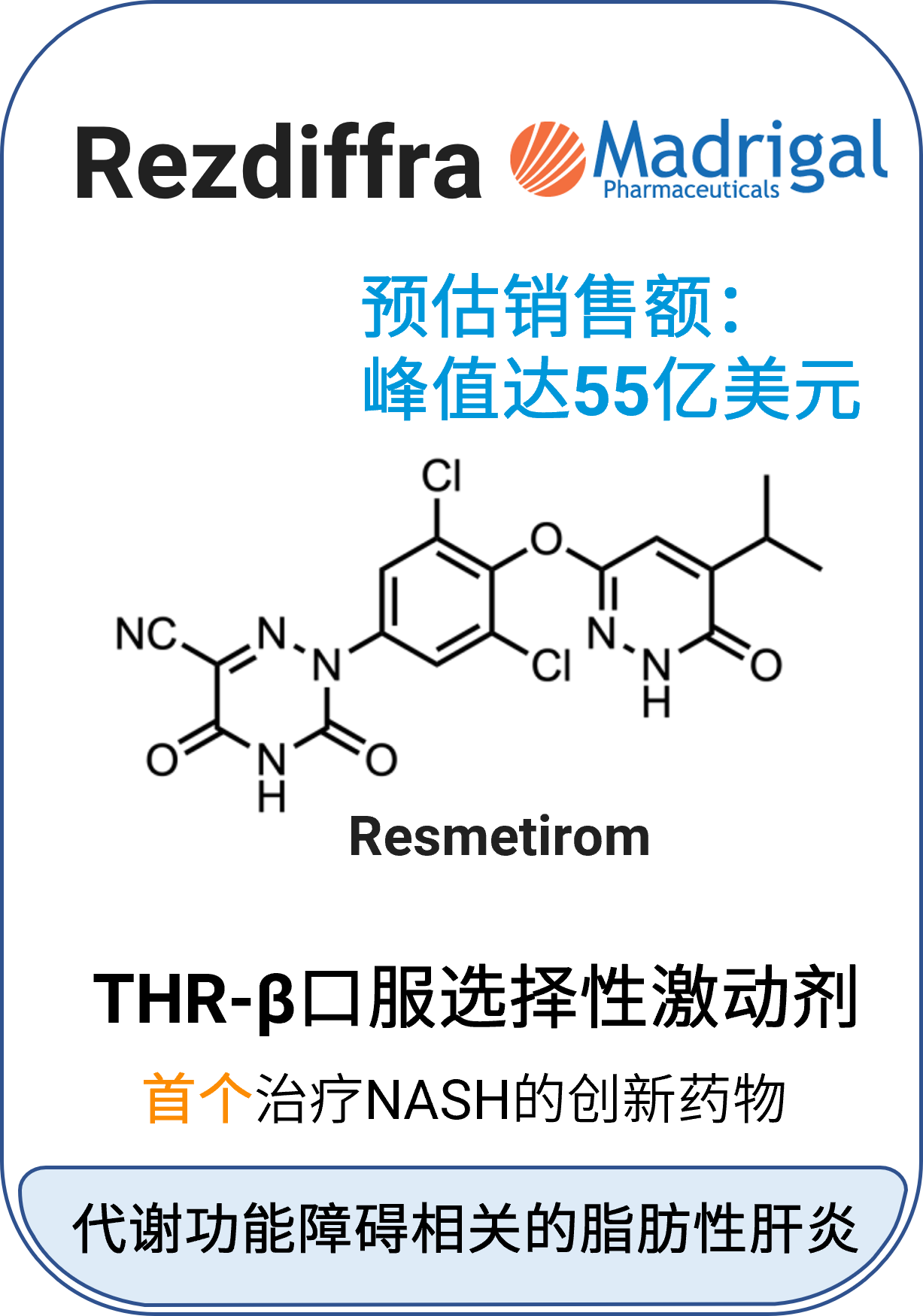
药代动力学及药物相互作用特征[5]:
吸收
每日一次给药,3至6天达到稳定状态;Cmax,ss: 778 ng/mL (41.5% CV)(80 mg/天给药);AUCtau,ss: 5850 ng·h/mL (60.5% CV)(80 mg/天给药);Cmax,ss: 971 ng/mL (40.9% CV)(100 mg/天给药);AUCtau,ss: 7780 ng·h/mL (65.5% CV)(100 mg/天给药);Tmax: 约4 h(80 mg/天或100 mg/天每日多次给药后);与空腹状态相比,伴随食物给药导致Cmax下降33%, AUC下降11%,Tmax延迟约2小时。
分布
分布容积: 68 L(227% CV);血浆蛋白结合率: >99%。
代谢
半衰期: 4.5小时;清除率: 17.5 L/h(56.3% CV);通过CYP2C8代谢,在体外不通过其他CYP酶代谢。
排泄
尿液: 24%;粪便: 67%。
药物相互作用
体外研究表明,resmetirom是CYP2C8、UGT1A4和UGT1A9的抑制剂,是OATP1B1、OATP1B3和BCRP的底物,抑制OATP1B1、OATP1B3、BCRP、OAT3和BSEP。
临床研究:
-
与clopidogrel(CYP2C8抑制剂)联用时,Cmax增加1.3倍、AUC增加1.7倍。
-
与pioglitazone(CYP2C8底物)联用时,使pioglitazone的AUC增加1.5倍。
-
与simvastatin(OATP1B1和OATP1B3底物)联用时,使simvastatin的Cmax增加1.4倍,AUC增加1.7倍。
-
与rosuvastatin(BCRP, OATP1B1和 OATP1B3 底物)联用时,使rosuvastatin的Cmax增加2.9倍,AUC增加1.8倍。
-
与pravastatin(OATP1B1和OATP1B3底物)联用时,使pravastatin的Cmax增加1.3倍,AUC增加1.4倍。
-
与atorvastatin(BCRP, OATP1B1和 OATP1B3 底物)联用时,使atorvastatin的AUC增加1.4倍,atorvastatin内酯Cmax增加2.0倍,AUC增加1.8倍。
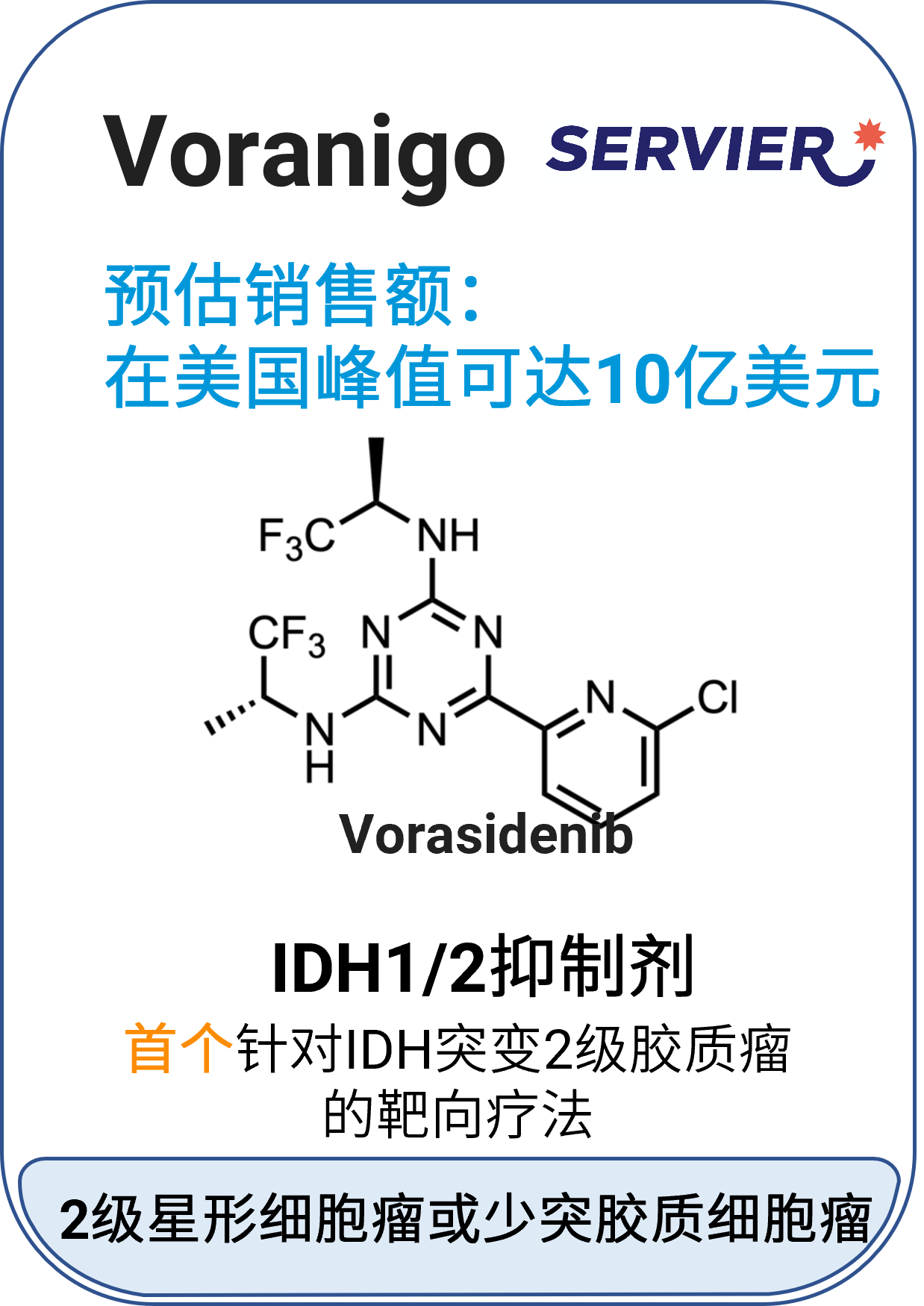
05 Voranigo™ (vorasidenib)
适应症:2级星形细胞瘤或少突胶质细胞瘤
作用机制:异柠檬酸脱氢酶-1和2(IDH1和IDH2)酶的靶向抑制剂
分子类型:小分子药物
获批日期:2024年8月6日
开发公司:Servier Pharmaceuticals
给药途径:口服
给药制剂:片剂
药代动力学及药物相互作用特征:
吸收
在10-200 mg的剂量范围内(最高批准推荐剂量暴露量的0.2至4倍),每日一次单次和多次给药后,暴露量大致成比例增加。在最高批准推荐剂量下,Cmax: 133 ng/mL (73% CV);AUC: 1988 h•ng/mL (95% CV);Tmax: 2小时(0.5至4小时);绝对生物利用度34%。食物效应:与空腹状态相比,高脂高卡路里饮食可提高Cmax 3.1倍,提高AUC 1.4倍;低脂低卡路里饮食可提高Cmax 2.3倍,提高AUC 1.4倍。
分布
分布容积: 3930 L (40% CV);血浆蛋白结合率: 97%;脑肿瘤-血浆浓度比: 1.6。
代谢
半衰期: 10天 (57% CV);清除率: 14 L/h (56% CV);主要通过CYP1A2代谢,非CYP途径可能占其代谢的30%。
排泄
粪便: 85%(56%原药);尿液: 4.5%。
药物相互作用
体外研究表明,vorasidenib是CYP2B6, CYP2C8, CYP2C9, CYP2C19和CYP3A以及UGT1A4诱导剂,是BCRP抑制剂。
与中强度CYP1A2抑制剂(如ciprofloxacin)联用时,AUC增加2.5倍;与CYP1A2诱导剂(如phenytoin)联用时,预计AUC减少40%;与强CYP1A2抑制剂(如fluvoxamine)联用时,预计暴露量增加≥5倍。
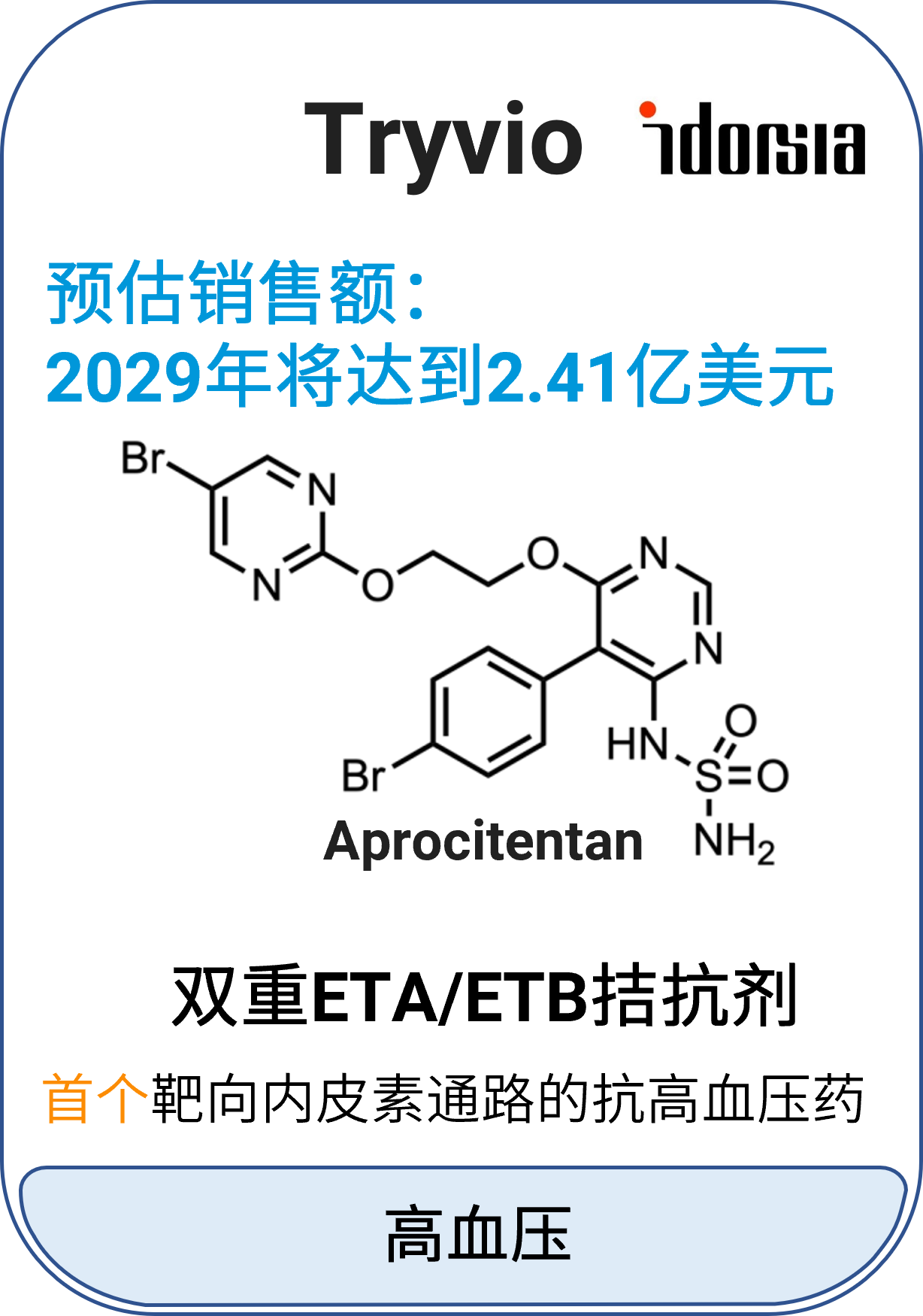
06 Tryvio™ (aprocitentan)
适应症:高血压
作用机制:双重内皮素(ET)A/B受体(ETA/ETB)拮抗剂
分子类型:小分子药物
获批日期:2024年3月19日
开发公司:Idorsia Pharmaceuticals
给药途径:口服
给药制剂:片剂
药代动力学及药物相互作用特征:
吸收
单次25 mg(推荐剂量的两倍)给药后,4-5小时达到Cmax;Cmax: 1.3 μg/mL (19% CV);AUC0-tau: 23 μg·h/mL (17% CV)。蓄积量为3倍(1次/天给药,8天后达到稳态浓度)。
分布
分布容积: 20 L;血浆蛋白结合率 >99%;全血-血浆分配比:0.63。
代谢
半衰期: 41 小时;清除率: 0.3 L/h;主要通过UGT1A1和UGT2B7介导的N-糖苷化和非酶水解代谢。
排泄
粪便: 25%(6.8%原药);尿液: 52%(0.2%原药)。
药物相互作用
体外研究表明:Aprocitetan抑制CYP3A4和整个CYP2C家族成员,诱导CYP3A4,是UGT1A1和UGT2B7的底物和抑制剂,是BCRP、BSEP和牛磺胆酸钠共转运多肽(NTCP)的抑制剂。
与UGT诱导剂联用时,aprocitentan暴露量减少。
07 Rytelo™ (imetelstat)
适应症:低至中度1级风险骨髓增生异常综合征
作用机制:寡核苷酸人端粒酶抑制剂,可与人端粒酶(hTR)RNA组分的模板区结合,抑制端粒酶酶活性并阻止端粒结合
分子类型:寡核苷酸药物
获批日期:2024年6月6日
开发公司:Geron Corporation
给药途径:静脉注射
给药制剂:注射液

药代动力学及药物相互作用特征:
吸收
在推荐剂量(7.1 mg/kg,静脉输注2小时以上,每4周一次)下,Cmax: 18.3 μM (27.3% CV%);AUC0-28天: 114.2 h·μM (43.2% CV%),在治疗周期之间不蓄积。
分布
分布容积: 14.1 L (27.2% CV%);血浆蛋白结合率: >94%。
代谢
半衰期: 4.9小时 (43.2% CV%);预计被核酸酶代谢为不同长度的核苷酸。
排泄
信息未提供。
药物相互作用
体外研究表明,imetelstat不是CYP450酶的抑制剂或诱导剂,但抑制OATP1B1和OATP1B3。
08 Tryngolza™ (olezarsen)
适应症:家族性乳糜微粒血症综合征
作用机制:一种ASO-GalNAc3结合物,与apoC-Ⅲ mRNA结合导致mRNA降解,血清apoC-Ⅲ蛋白减少,导致血浆TG和VLDL清除率增加
分子类型:寡核苷酸药物
获批日期:2024年12月19日
开发公司:Ionis Pharmaceuticals
给药途径:皮下注射
给药制剂:注射液

药代动力学及药物相互作用特征:
吸收
在获批的患者推荐剂量(80 mg,每月一次)下,Cmax: 883 ng/mL;AUCtau: 7440 ng·h/mL;Tmax: 约2小时(皮下给药后);重复给药时无蓄积。
分布
中央分布容积: 91.9 L;外周分布容积: 2960 L;血浆蛋白结合率: >99%;主要分布于肝脏和肾脏。
代谢
半衰期: 约4周;主要在肝脏内由内切核酸酶和外切核酸酶代谢为不同大小的寡核苷酸片段。
排泄
尿液中少于1%的剂量在24小时内以原型排出。
药物相互作用
体外研究表明:Olezarsen不是CYP450酶的抑制剂或诱导剂;不是OAT1、OAT3、OCT1、OCT2、OATP1B1、OATP1B3、MATE1、MATE2-K、BCRP、P-gp和BSEP的底物或抑制剂;不置换华法林和布洛芬的血浆蛋白结合位点。
09 Tevimbra™ (tislelizumab-jsgr)
适应症:不可切除或转移性食管鳞状细胞癌
作用机制:程序性死亡受体-1(PD-1)阻断抗体
分子类型:单克隆抗体
获批日期:2024年3月13日
开发公司:BeiGene
给药途径:静脉注射
给药制剂:注射液

药代动力学特征:
吸收
在批准的推荐剂量(200 mg静脉滴注,每3周1次)下,Cmax: 110 µg/mL (22.2% CV);AUCtau: 1283 µg/mL·day(28.7% CV);蓄积量为2.14倍(1次/3周给药,12周后达到稳态浓度)。
分布
分布容积: 6.42 L (32.6% CV)。
代谢
半衰期: 24天(31% CV);清除率: 0.153 L/天(29.5% CV)。
排泄及药物相互作用
信息未提供。
10 Kisunla™ (donanemab-azbt)
适应症:阿尔茨海默病
作用机制:β-淀粉样蛋白靶向抗体
分子类型:单克隆抗体
获批日期:2024年7月2日
开发公司:Eli Lilly
给药途径:静脉注射
给药制剂:注射液

药代动力学特征:
吸收
推荐剂量为700 mg,静脉输注,每4周一次,随后为1400 mg,每4周一次。无蓄积:每4周给药一次,蓄积<1.3倍,单次给药后即达到稳态,剂量依赖性: 暴露量在10到40 mg/kg单剂量和10到20 mg/kg多剂量范围内成比例增加。
分布
中央分布容积: 3.36 L。
代谢
半衰期: 12.1天,清除率: 0.0255 L/h,预计由蛋白水解酶降解,类似内源性IgG。
排泄
预计不通过肾脏消除。
药物相互作用
信息未提供。
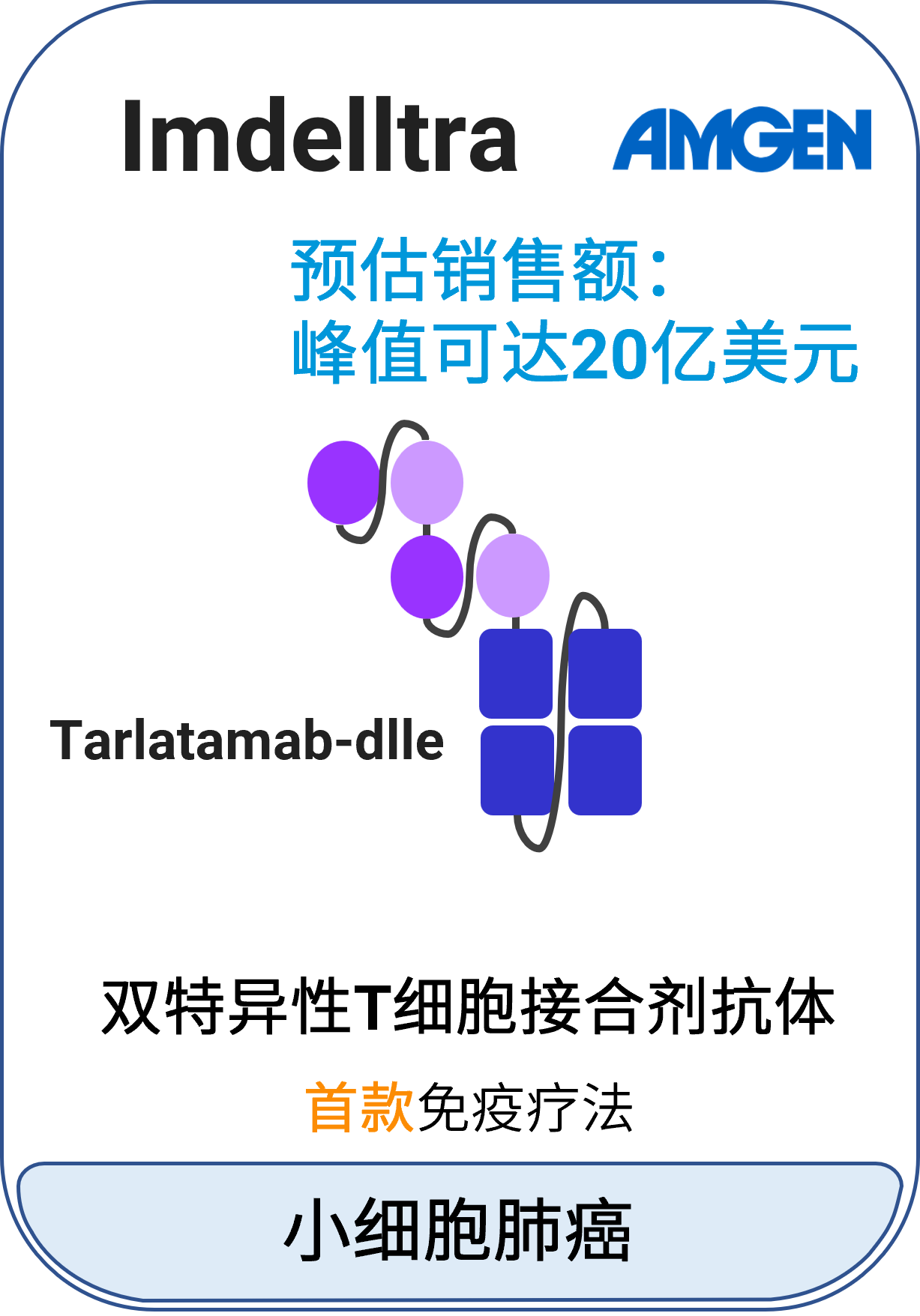
11 Imdelltra™ (tarlatamab-dlle)
适应症:广泛期小细胞肺癌
作用机制:双特异性T细胞衔接器,与细胞表面表达的DLL3(包括肿瘤细胞)和T细胞表面表达的CD3结合,引起T细胞活化、炎性细胞因子释放和DLL3表达细胞裂解
分子类型:双特异性抗体药物
获批日期:2024年5月16日
开发公司:Amgen
给药途径:静脉注射
给药制剂:注射液
药代动力学及药物相互作用特征:
吸收
在1 mg至100 mg每2周一次(最高获批推荐剂量的10倍)剂量范围内,tarlatamab-dlle暴露量与剂量成比例增加。第2周期第15天达到稳态暴露量::Cavg: 1040 ng/mL (44% CV);Cmax: 3400 ng/mL (40% CV);Ctrough: 495 ng/mL (73% CV)。
分布
分布容积: 8.6 L (18.3% CV)。
代谢
在小细胞肺癌(SLCL)患者中,清除率: 0.65 L/天 (44% CV),半衰期:11.2天(4.3至26.5天)。预计通过蛋白质代谢途径降解为小肽。
排泄
信息未提供。
药物相互作用
可引起细胞因子释放,可能抑制CYP450酶,导致同时使用的CYP底物暴露量增加。
12 Winrevair™ (sotatercept-csrk)
适应症:肺动脉高压
作用机制:激活素信号传导抑制剂,可改善促增殖(ActRIIA/Smad2/3介导)和抗增殖(BMPRII/Smad1/5/8介导)信号之间的平衡,以调节血管增殖
分子类型:同源二聚体重组融合蛋白
获批日期:2024年3月26日
开发公司:Acceleron Pharma & Merck
给药途径:皮下注射
给药制剂:注射液

*文中图片的药物预估销售额数据均来自于参考文献6。
药代动力学特征:
吸收
每3周注射0.7 mg/kg,约15周后达到稳态;AUC: 172 μg·d/mL (34.2% CV); Cmax: 9.7 μg/mL(30% CV);AUC蓄积比约2.2;皮下注射后绝对生物利用度:约66%;每4周多次皮下注射给药,Tmax约为7天。
分布
分布容积: 5.3 L(27.3% CV)(肺动脉高压患者);中心分布容积随体重增加而增加。
代谢
半衰期: 24天;清除率: 0.18 L/day;清除率随体重增加而增加,预期通过分解代谢途径代谢为小肽。
排泄及药物相互作用
信息未提供。
结 语
通过分析2024年FDA获批的几种典型新药的药代动力学和药物相互作用特性,我们可以发现,不同分子类型和适应症的药物在吸收、分布、代谢和排泄方面都有各自的特点和优势。这些新药在药代动力学和药物相互作用特性上的明显优化,为未来药物开发提供了宝贵的经验和参考。以Rezdiffra(Resmetirom)为例,其开发过程体现了ADME研究从“被动优化”向“主动设计”的转变。这种转变的核心在于,通过早期的分子结构设计,系统性地整合了ADME的各个特性。在吸收方面,通过优化分子结构中的亲脂性和极性平衡,提高了药物的吸收效率。在分布方面,依赖肝特异性转运蛋白OATP1B1实现了药物在肝脏中的靶向分布。在代谢方面,通过主动规避有毒代谢物的生成,并增强了代谢稳定性。在排泄方面,主要通过肝胆排泄,降低了肾毒性风险,满足了慢性肝病长期用药的需求。
药明康德DMPK团队具备丰富的经验,涵盖小分子、生物药、核酸多肽等多个领域,并在ADME筛选和优化方面具有深厚的专业知识,能够助力客户更快地推进项目到临床阶段,同时帮助发现和规避临床中可能出现的风险。
作者:潘岩,颜昀昳,彭婷,王宇,侯丽娟,金晶
编辑:钱卉娟,富罗娜·克里木
设计:倪德伟,张莹莹
药明康德DMPK依托中国(上海、苏州、南京和南通)和美国(新泽西)的研发中心,提供从早期筛选、临床前开发、到临床研究阶段的综合型药代动力学服务,助力您快速推进药物研发流程。拥有上千人的研发团队,服务超1600家全球客户,具有超过十五年的新药申报经验,已成功支持超过1700个新药临床研究申请(IND)。
点击此处与我们的专家进行联系。
参考
参考文献:
[1] FDA.Novel Drug Approvals for 2024. http://www.fda.gov/drugs/novel-drug-approvals-fda/novel-drug-approvals-2024.
[2] Mullard, A. 2024 FDA approvals. Nat Rev Drug Discov 24, 75–82 (2025).
[3] de la Torre, B. G. & Albericio, F. The Pharmaceutical Industry in 2024: An Analysis of FDA Drug Approvals from the Perspective of Molecules. Preprint at http://doi.org/10.20944/preprints202501.0253.v1 (2025).
[4] FDA & CDER. HIGHLIGHTS OF PRESCRIBING INFORMATION. www.fda.gov/medwatch.
[5] FDA. CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 217785Orig1s000 INTEGRATED REVIEW.
[6] 2024 drug approvals: Small companies loom large with several key FDA nods. http://www.fiercepharma.com/pharma/2024-drug-approvals.














加入订阅
获取药物代谢与药代动力学最新专业内容和信息

