





肽是α-氨基酸以肽键连接在一起而形成的化合物。2-20个氨基酸组成的肽称为寡肽,更多个氨基酸分子组成叫多肽。分子量一般在10 kDa以下,其与蛋白质并没有明确的界限。根据FCA (Further Consolidated Appropriations Act, 2020) 对BPCIA (Biologics Price Competition and Innovation Act of 2009) 的修订,FDA 将术语“蛋白质”解释为“具有明确定义的序列且大小大于40个氨基酸的任何α氨基酸聚合物”,属于生物制品许可申请(BLA)[1]。因此小于等于40个氨基酸的多肽药物研发仍可参考小分子的监管要求。

图1. 氨基酸、多肽与蛋白结构
我们知道生命活动主要基于蛋白质之间的相互作用,而蛋白质结构或功能的变化会引发疾病产生。多肽作为蛋白质的结构单元对疾病的治疗当然也起到了关键的作用。
Nature Review Drug Discovery上发表的综述文章”Trends in peptide drug discovery”[2]介绍了多肽药物的研究历史和趋势。1922年,F.G. Banting和C.H. Best从牛胰腺中提取的胰岛素用于治疗1型糖尿病,成为第一个多肽类药物,开启了多肽类药物研发的历史。1954年,Vincent du Vigneaud的团队首次用化学合成多肽,发表了”The total synthesis of oxytocin and vasopressin”并在1955年获得诺贝尔化学奖。下一个飞跃是Bruce Merrifield在固相上组装氨基酸以自动合成多肽的想法,导致了1963年固相肽合成(SPPS)的发明,并于1984年获得诺贝尔化学奖。20世纪80年代重组技术的出现使更大的多肽的清洁生产成为可能。
历经这些多肽研究的里程碑,再加上二十世纪后半段新药研发的浪潮,到2020年已有超过80个多肽药物上市,多肽类药物已成为医药行业最具有广泛市场前景的研发发展方向之一。
一、多肽药物的特征与优势
多肽类药物兼具了小分子药物和蛋白质药物的优势。小分子与靶点蛋白结合面小、活性低、特异性差,容易导致毒副作用。多肽与靶点亲和力强、特异性高、副作用小[3]。与大分子药物相比,多肽药物具有免疫原性低、生产成本低等明显优势。然而,多肽药物开发面临渗透性低、稳定性较差、口服生物利用度低等挑战[4]。

图2. 多肽药物的特征与优势

表1. 各类药物的优缺点比较[5]
二、多肽的DMPK性质
Absorption 吸收
多肽吸收主要方式为液体的对流、被动扩散和受体介导的主动转运。除寡肽或小肽外,多肽的分子量通常大于1000 Da且极性较大,因此被动扩散较小分子缓慢。分子量较大的多肽(5000~12 000 Da)主要通过淋巴吸收和转运。由于淋巴液循环较慢,药物的达峰时间会显著延长。体内药代还容易因为吸收迟滞产生Flip-flop现象[6](吸收常数远小于消除常数),导致半衰期和药效作用的延长。例如促性腺激素释放激素(GnRH)激动剂亮丙瑞林,其每月一次的长效肌肉注射剂会缓慢释放药物到系统循环中,以长期抑制促卵泡激素(FSH)和促黄体激素(LH)用于治疗前列腺癌和子宫内膜异位。
口服多肽药物会遇到胃肠道黏液层,该层结构分泌一些黏液蛋白,通过分子间相互作用和二硫键阻碍多肽类药物向细胞的扩散。Kovalainen等报道,肠上皮细胞的渗透仅限于相对分子质量≤3500 Da的分子[7]。所以胃肠道吸收差成为口服多肽药物开发的一个难点,要提高多肽的吸收常会采取以下一些办法。
根据药物的酸碱性、分子量、LogP等特征找寻合适的吸收位点,采取相应的制剂手段延长在吸收部位的停留时间;
尝试合适的吸收促进剂(如表面活性剂、胆盐、磷脂、脂肪酸、甘油酯等),增加胃肠道的通透性。另外癸酸钠(C10,又称癸酸)和钠N-[8-(2-羟基苯甲酰基)氨基]辛酸酯(SNAC,又称沙卡普罗酸钠)也是常用的渗透增强剂;
采用生物粘附的制剂,比如Polycarbophil,提高在黏液层中的吸收。
Distribution 分布
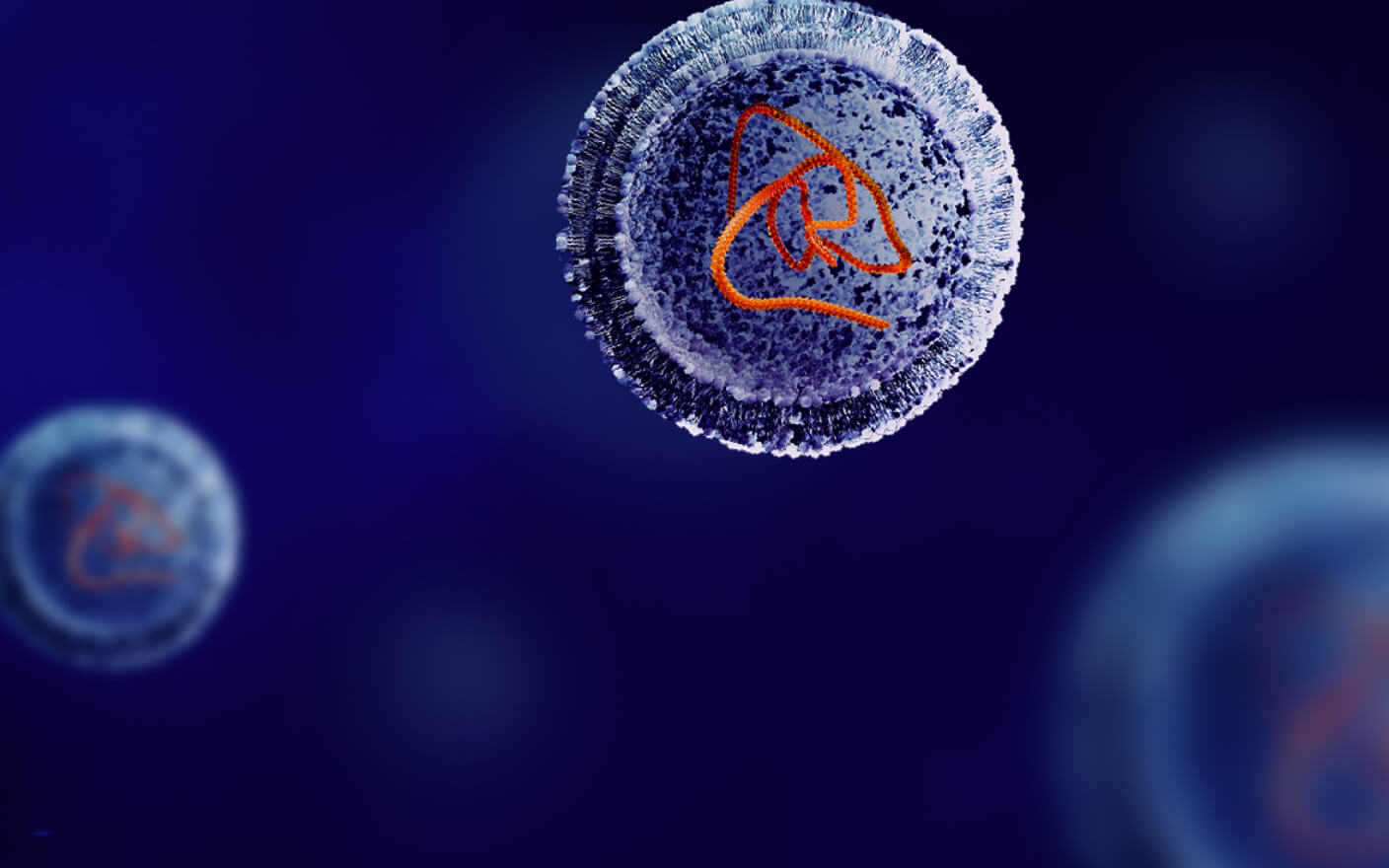
人血清白蛋白(human serum albumin,HSA)与多肽的结合能力强,生理浓度高,对于多肽的分布有较大影响。多肽的亲脂性弱、渗透性低,较难直接穿过细胞膜进入细胞。而主要采用胞吞作用进入细胞,包括胞饮或者吞噬作用,有时要与一些细胞膜表面受体结合才能进入细胞。因为多肽的Vd值通常较小,一般不大于细胞外液的体积,为了促进多肽的细胞内递送和靶向作用,采用包括脂质体、聚合物、蛋白质复合物、无机材料等在内的各种纳米载体系统被认为是较好的策略。
还有一种特殊的多肽为穿膜肽(Cell-Penetrating Peptide, CPP),它是一类可携带分子或蛋白进行透膜的短肽,主要来自于病毒衍生的肽序列,非病毒蛋白质或较小的分子。其穿膜能力不依赖经典的胞吞作用,能够直接穿过细胞膜进入细胞。

图3. 多肽进入细胞的几种方式[8]
Metabolism 代谢
多肽主要通过蛋白酶或肽酶进行降解。蛋白酶在机体内广泛分布,包括肝、肾和胃肠道组织,肺、血液和血管内皮、皮肤及其他组织和器官。蛋白酶分为内肽酶和外肽酶(包括氨肽酶和羧肽酶)[9]。肽和蛋白质的水解通常起始于内肽酶,它作用于蛋白质的中间部分,酶解产生的寡肽可进一步被外肽酶降解。一些多肽的前药或者是代谢产物也可以被熟知的CYP450进行代谢。

表2. 代谢多肽的蛋白酶及其代谢位点
除了被血浆、胃肠道中的蛋白酶等直接代谢外,细胞内吞消除和靶点介导的消除(Target-mediated drug disposition, TMDD)[10]也是多肽的主要消除途径。

表3. 细胞内吞消除及靶点介导的药物消除途径比对
为了提高多肽化合物的稳定性,可以从以下几个方面进行优化[11]:
N-或C-端修饰,如甲基化、N端乙酰化。人肠促胰岛素葡萄糖依赖性促胰岛素肽(GIP)(1-42)的t1/2很短只有2-5分钟,Mabilleau等人通过乙酰化GIP中的Tyr1(N-AcGIP)开发了一种酶促稳定的GIP类似物,循环t1/2大于24小时;
用D-氨基酸和人造氨基酸替代天然氨基酸、降低与蛋白酶的亲和力;
环化或者双环化,提高化合物刚性;
增加分子量,如添加脂肪链、PEG化;
添加特异性配体提高与白蛋白的结合,可以有效延长多肽药物半衰期。
Excretion 排泄
肾小球孔隙约为8 nm,一般认为小于50 kDa的分子均可被肾脏滤过作用清除[12],多肽分子越小,肾清除率越大。肾功能受损有可能缩短多肽类药物的半衰期。
经肾小球滤过的较大的多肽,通过胞吞和溶酶体降解清除,最后水解成小肽碎片和氨基酸。经肾小球滤过的较小的多肽被近端肾小管腔内刷状边缘膜中的肽链端解酶水解成氨基酸,再经特异性氨基酸转运系统被重新吸收进入体循环;或是先断裂成小肽,再转运至近曲小管上皮细胞内,在胞内水解。

表4. 多肽与其他各类药物的DMPK性质区别
三、多肽化合物DMPK研究策略
药明康德药物代谢与动力学部多年来已经完成数百个多肽化合物的研究项目,在研究策略、实验设计、样品分析、问题解决、数据解读等方面积累了丰富的经验,建立了完善的多肽成药性评价体系。在筛选阶段,需要考察血浆稳定性、血浆蛋白结合率、肾脏匀浆液稳定性实验。多肽药物的排泄实验常采用同位素标记的方法。在IND申报阶段,要充分考虑药物在各代谢体系中的种属差异,选择与人代谢特征相近的动物种属;依据药理机制与清除特征,尤其是可能的非线性动力学,确定科学的给药方式、剂量与检测时间点。由于多肽药物可能有免疫原性,需要检测抗药抗体(ADA)的浓度以评估可能出现的药理学影响。

表5. 多肽药物不同研发阶段的药代动力学研究内容
在临床数据预测方面,不同的哺乳动物对肽类的处理方式相对比较保守,这就意味着根据动物身上获得的药代动力学数据来推测人类中的情形具有相当高的可靠性。因此使用异速放大法进行预测往往是比较准确的。但对于分子量比较小的多肽,肝代谢为主要的清除途径或种属间代谢差异比较大时,可能IVIVE是更合适的方法,具体可以点击查看我们此前发布的专题文章:IVIVE代谢预测模型助力药物研发:如何利用体外药物代谢数据推导其体内的代谢清除特征。
结语
多肽药物具有靶点丰富、生物活性高、特异性强、副作用小的优点。近年来,人们对多肽药物的关注度日渐提高。虽然多肽药物的开发存在较多难点,但不断出现的新型生物合成、制剂和递送载体等手段将会给多肽药物的应用带来巨大突破。与其它类型药物的结合的如PDC也具有非常广阔的前景。多肽药物在制药领域中占据了一个明确的空间,它将可能凭借独特的优势超越小分子和大分子生物制剂,给全球病患带来健康福音。
药明康德DMPK依托在中国(上海、苏州、南京和南通)和美国(新泽西)的研发中心,提供从早期筛选、临床前开发、到临床研究阶段的综合型药代动力学服务,助力您快速推进药物研发流程。拥有上千人的研发团队,服务超1500家全球客户,具有超过十五年的新药申报经验,已成功支持超过1200个新药临床研究申请(IND)。
点击此处可与我们的专家进行联系。
作者:孙建平,潘岩,王宇,金晶
编辑:方健,钱卉娟
设计:倪德伟
参考
[1] ANDAs for Certain Highly Purified Synthetic Peptide Drug Products That Refer to Listed Drugs of rDNA Origin, Guidance for Industry, Food and Drug Administration
[2] Muttenthaler, M., King, G.F., Adams, D.J. et al. Trends in peptide drug discovery. Nat Rev Drug Discov (2021). https://doi.org/10.1038/s41573-020-00135-8
[3] Ved Srivastava, Peptide-based Drug Discovery: Challenges and New Therapeutics
[4] Lau, Jolene L. and M. K. Dunn. “Therapeutic peptides: Historical perspectives, current development trends, and future directions.” Bioorganic & medicinal chemistry 26 10 (2018): 2700-2707 .
[5] Sara La Manna, Peptides as Therapeutic Agents for Inflammatory-Related Diseases. 2018 Int. J. Mol. Sci 19 2714; Philippe 2021 Drug Discovery
[6] Di, L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J 17, 134–143 (2015). https://doi.org/10.1208/s12248-014-9687-3
[7] 丁海波等,多肽类药物药代动力学特点及其代谢机制研究进展,Chin J Pharmacol Toxicol,Vol 32,No 3,Mar 2018,DOI:10.3867/j.issn.1000-3002.2018.03.010
[8] Neeraj Tiwari Heidelberg, Univ., Diss., 2005 (Nicht für den Austausch).
[9] 姚金凤等,多肽类药物代谢研究进展,Chinese Pharmacological Bulletin 2013 Jul; 29(7): 895-899
[10] Peletier, L.A. & Gabrielsson, J. Dynamics of target-mediated drug disposition: characteristic profiles and parameter identification. J Pharmacokinet Pharmacodyn (2012) 39: 429. https://doi.org/10.1007/s10928-012-9260-6
[11] Gaurang Patel, Ambikanandan Misra, Oral Delivery of Proteins and Peptides: Concepts and Applications
[12] Amita Datta-Mannan.(2019) Mechanisms Influencing The Pharmacokinetics And Disposition Of Monoclonal Antibodies And Peptides. Drug Metabolism and Disposition October 2019, 47 (10) 1100-1110; DOI: https://doi.org/10.1124/dmd.119.086488














加入订阅
获取药物代谢与药代动力学最新专业内容和信息

