

Antibody-Drug Conjugate (ADC) R&D Innovations and In Vitro ADME Research Considerations
-

Articles
-

Jul 24, 2025
In April 2024, a Nature Reviews Drug Discovery article titled "The Antibody–Drug Conjugate Landscape" highlighted that antibody-drug conjugates (ADCs) have emerged as a key treatment modality in oncology. Compared to conventional chemotherapy across multiple indications, ADCs demonstrate superior clinical efficacy. Consequently, the combined revenue of approved ADC drugs and those in Phase III trials is projected to reach $26 billion by 2028 [1]. Despite the approval of 15 ADCs, two major challenges persist: first, the limited mechanisms of action of current payloads restrict their applicability; second, nonspecific and inefficient payload delivery narrows the therapeutic window. This article summarizes innovations in ADC design across three key components—targets, payloads, and linkers—and reviews experimental platforms for in vitro ADME studies in ADC research.
What are Antibody-Drug Conjugates?
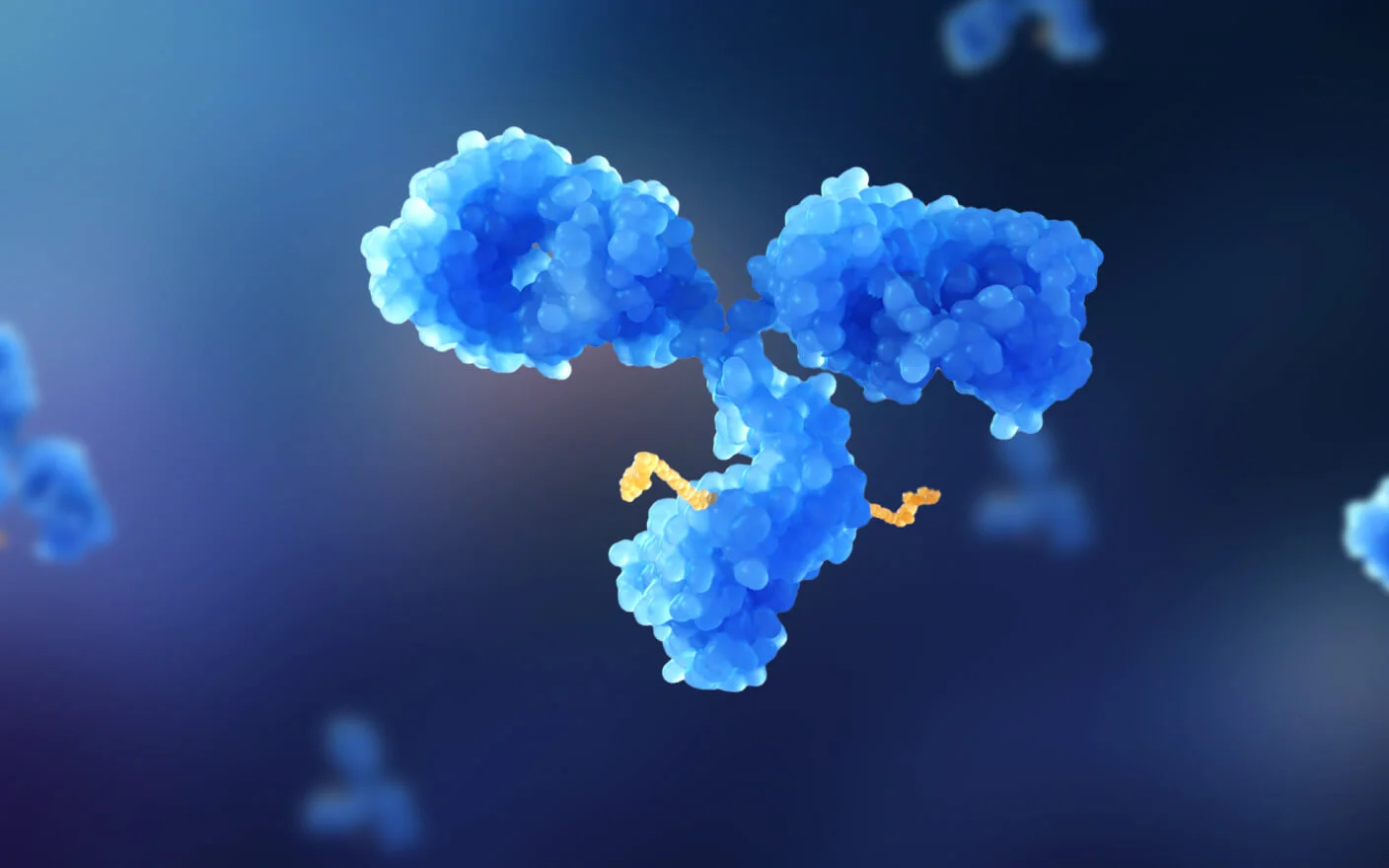
Antibody-drug conjugates (ADCs) are a class of targeted biopharmaceuticals that deliver cytotoxic agents to tumor cells by covalently linking a monoclonal antibody to a potent small-molecule payload through a synthetic linker. The design of ADCs demands:
An antibody with high antigen-binding affinity and minimal off-target binding.
A linker stable in systemic circulation yet cleavable in the tumor microenvironment.
A payload with sub-nanomolar cytotoxicity and suitable pharmacokinetic properties.
The ideal ADC maintains stability in blood, precisely targets tissues, and releases cytotoxins locally.
: antibody, linker, and payload")
Figure 1. Structural components of antibody-drug conjugates (ADCs): antibody, linker, and payload [2].
Target Innovations
Current ADC development has predominantly focused on established targets such as HER2 and TROP2. Analysis reveals that approximately 90% of ADC targets are tumor-associated antigens overexpressed on cancer cells, while the remaining 10% target unique tumor microenvironment features. A promising novel target is the extra domain B (EDB) of fibronectin (FN), a high molecular weight extracellular matrix protein. While FN is ubiquitously expressed in normal tissues, its EDB domain demonstrates significant tumor-specific overexpression.

Figure 2. Differences in the expression of EDB+FN protein in tumor stroma and normal tissues[3].
Figure 2 demonstrates this differential expression pattern between tumor stroma and normal tissues. The immunohistochemical analysis shows marked overexpression in tumor stroma (purple frame) compared to minimal expression in normal thyroid and liver tissues (green frame). The therapeutic potential of targeting FN-EDB is exemplified by PYX-201, an investigational ADC that demonstrated promising efficacy data in presentations at the 2024 American Association for Cancer Research (AACR) Annual Meeting [1].
Small-Molecule Payload Innovations
Current ADC payloads primarily employ three mechanistic classes: (1) antimitotic agents, (2) topoisomerase I inhibitors, and (3) DNA alkylating agents. Established warheads include MMAE (a microtubule-disrupting agent) and DXd (a topoisomerase I inhibitor). An emerging paradigm involves small-molecule degraders targeting GSPT1, a translation termination factor frequently overexpressed in malignancies. These degraders demonstrate:
Picomolar potency
High target specificity
Polypharmacology against cancer-related proteins
Apoptosis induction via GSPT1 degradation
The investigational ADC ORM-5029 incorporates such a GSPT1-targeting degrader. Preclinical studies in breast cancer models revealed antitumor activity comparable to trastuzumab deruxtecan (Enhertu®)[1].
Linker Innovations
Common linkers include dipeptides like VC and tetrapeptides like GGFG, which can be cleaved by cathepsins. Emerging linker technologies focus on the exogenous control of payload release rather than relying on endogenous enzyme-mediated cleavage. The principle involves ADC binding to extracellular tumor targets, followed by the intravenous injection of an exogenous chemical probe (activator) that rapidly reacts with the ADC linker to release the drug, bypassing dependence on tumor biology. The ADC drug TGW101, in development, uses an exogenous chemical activator to induce payload release, showing superior anti-tumor activity in colorectal and ovarian cancer xenograft models compared to VC dipeptide linkers[1].
Innovations in these three areas demonstrate the broad development potential of antibody-drug conjugates. Many drugs in development focus on traditional designs combined with new small-molecule toxins or linkers, blending classic and innovative approaches to create more rational antibody-drug conjugates.
In Vitro ADME Studies of Antibody-Drug Conjugates
Based on the in vivo processes of antibody-drug conjugates, WuXi AppTec DMPK has established corresponding testing platforms. After intravenous injection, ADC drugs must remain stable in the circulatory system. To assess the stability of antibody-drug conjugates, plasma/whole blood study systems can be used. Upon reaching the target tissue, antibodies specifically bind to antigens, are internalized into tumor cells, and linkers are cleaved in lysosomes, releasing cytotoxins that induce apoptosis. To evaluate the mechanism of cytotoxin release, lysosomes, cathepsin B, acidified liver homogenate, and liver S9 test systems have been established. Additionally, the release capacity and the cytotoxicity of cytotoxins in tumor target cells can be evaluated in vitro. Through these assessments, the stability in the circulatory system and the mechanism of toxin release can be better understood, leading to more rational ADC drug design.
Plasma Stability Studies of ADCs
ADC drug DS8201 was selected for study, with GGFG and Dxd as its linker and payload, respectively. GGFG is a commonly used linker, and Dxd's mechanism is topoisomerase I inhibition.
Table 1. DS8201 Basic Information
Drug Name | Target | Linker | Payload | DAR Value |
DS8201 | HER2 | GGFG | Dxd | 8 |
By incubating DS8201 with plasma from different species and PBS containing 1% BSA for various time points up to 21 days, the concentration of Dxd released in plasma was measured, and its release rate was calculated. The data are shown below:

Fgure 3. Dxd Release Concentration in Plasma and PBS containing 1% BSA from Different Species
Table 2. Dxd Release Concentration and Release Rate after 21 Days of Incubation with DS8201 and Plasma
ADC Incubation Concentration | Incubation Time (days) | Dxd Release Concentration (ng/mL) | Dxd %Release | Source | ||
Rat | Human | Rat | Human | |||
0.1 mg/mL | 21 | 22.8 | 22.9 | 0.88 | 0.89 | In-house |
42.8 | 37.4 | 1.66 | 1.45 | Literature [4] | ||
0.01 mg/mL | 2.8 | 1.8 | 0.11 | 0.07 | In-house | |
4.6 | 5.1 | 0.18 | 0.20 | Literature [4] | ||
Figure 3 shows the payload release concentration of DS8201 in plasma from different species and the negative control group. The release rate is less than 2%, indicating stability in the circulatory system, consistent with the design concept of ADC drugs. The difference in Dxd release concentration after 21 days of incubation and the reported material is within threefold, suggesting the plasma system is suitable for assessing long-term ADC stability. Considering that Dxd has two forms under different pH conditions, this system should be further expanded and optimized.
Payload Release Studies of ADC Drugs in Different Test Systems
Brentuximab vedotin is an antibody-drug conjugate launched in 2020, targeting CD30 with a classic VC dipeptide linker and MMAE toxin.
Table 3. Basic Information of Brentuximab Vedotin
Drug Name | Target | Linker | Payload | DAR Value |
Brentuximab Vedotin | CD30 | Val-Cit | MMAE | 4 |
By incubating with cathepsin B, liver lysosomes, liver homogenate, and liver S9 for different time points, the concentration of MMAE released was measured, and its release rate was calculated. The data is shown below:

Figure 4. MMAE Release Concentration in Different Matrices
Figure 4 shows the release concentration curves of MMAE after incubation in different test systems. In cathepsin B and liver homogenate, the release amount is similar, while liver lysosomes and liver S9 show different degrees of release. When exploring the payload release mechanism in ADC drugs, different test systems can be chosen based on the linker type (dipeptide, tetrapeptide, glycosidic bond, etc.) to design more appropriate linkers. For example, cathepsin B, a single enzyme, cleaves specific linkers (VC, GGFG, etc.), so those linkers can be directly studied with cathepsin B. Lysosomes contain multiple hydrolases that can hydrolyze exogenous and endogenous macromolecules, making them a preferred choice for linkers with unclear cleavage mechanisms.
Concluding Remarks
Opportunities and challenges coexist in the development of antibody-drug conjugates (ADCs). The future development of ADC drugs will move towards dual-target ADCs, dual-payload ADCs, and novel linker directions. With advancements in small-molecule screening and protein recombination molecular biology techniques, new conjugation modes in drug development have emerged, including antibody fragment-cytotoxin conjugates and antibody-oligonucleotide conjugates (AOCs). WuXi AppTec DMPK has established various research platforms to evaluate ADC drug stability in the circulatory system and payload release mechanisms. Facing the era of "XDC", WuXi AppTec DMPK embraces challenges and continuously enriches in vitro experiments, establishing more assessment platforms such as cathepsins L/K and glucuronidase to study linker cleavage efficiency, better serving the ADC drug development.
Authors: Haijuan Liu, Xiangling Wang, Genfu Chen
Talk to a WuXi AppTec expert today to get the support you need to achieve your drug development goals.
Committed to accelerating drug discovery and development, we offer a full range of discovery screening, preclinical development, clinical drug metabolism, and pharmacokinetic (DMPK) platforms and services. With research facilities in the United States (New Jersey) and China (Shanghai, Suzhou, Nanjing, and Nantong), 1,000+ scientists, and over fifteen years of experience in Investigational New Drug (IND) application, our DMPK team at WuXi AppTec are serving 1,600+ global clients, and have successfully supported 1,700+ IND applications.
Reference
[1] Patrick Flynn, Smruthi Suryaprakash, Dan Grossman, Val Panier & John Wu, The antibody–drug conjugate landscape. Nature Review Drug Discovery, 2024.
[2] Siler Panowski, et al, Site-specific antibody drug conjugates for cancer therapy. mAbs, 2014
[3] Sharon Wilks, et al, A First-in-Human Phase 1 Clinical Study Evaluating Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Efficacy of the EDB+FN targeting ADC PYX-201 in Participants with Advanced Solid Tumors. Poster.
[4] NDA/BLA Multi-disciplinary Review and Evaluation. Center for Drug Evaluation and Research, 2019, Enhertu.







Related Resources


Related Services and Platforms


-
 In Vitro ADME ServicesLearn More
In Vitro ADME ServicesLearn More
-
Novel Drug Modalities DMPK Enabling PlatformsLearn More
-
Physicochemical Property StudyLearn More
-
Permeability and Transporter StudyLearn More
-
Drug Distribution and Protein Binding StudiesLearn More
-
Metabolic Stability StudyLearn More
-
Drug Interactions StudyLearn More
-
PROTAC DMPK ServicesLearn More
-
ADC DMPK ServicesLearn More
-
Oligo DMPK ServicesLearn More
-
PDC DMPK ServicesLearn More
-
Peptide DMPK ServicesLearn More
-
mRNA DMPK ServicesLearn More
-
Covalent Drugs DMPK ServicesLearn More





Stay Connected
Keep up with the latest news and insights.

