

Exploring T-Cell Engagers (TCEs): Mechanism, Challenges, and Pharmacokinetic Considerations
-

Articles
-

Jul 04, 2025
T-cell-based immunotherapies, leveraging adaptive immunity, have revolutionized the cancer treatment landscape since their emergence. These therapies effectively eliminate malignant cells by targeting and mobilizing T cells while restoring and enhancing their anti-tumor activity. Based on their mechanisms of action, T cell-based cancer immunotherapies are primarily classified into two categories[1]: The first category targets immune checkpoint molecules such as PD-1 and PD-L1, with immune checkpoint inhibitors being the representative therapy. These inhibitors were among the first successful immunotherapies to restore T-cell anti-tumor activity, dating back to 2011 when ipilimumab received FDA approval for treating metastatic melanoma [2]. However, these therapies benefit only a subset of patients [3], spurring the development of the second category focusing on immune-stimulating pathways.
The second category is exemplified by chimeric antigen receptor (CAR) T cells and T-cell engagers (TCEs). While CAR-T cell therapy offers advantages such as high specificity and personalized treatment, its broad application is limited by high manufacturing costs, relatively long production cycles, and impaired T-lymphocyte quantity and function post-chemotherapy. In contrast, T-cell engagers have emerged as a promising cancer immunotherapy that has captured the attention of drug developers. This review will discuss the recent advances in T-cell engagers as well as their pharmacokinetic (PK) strategies and properties.
What are T-Cell Engagers and Their Mechanism of Action
The design of bispecific antibodies (BsAbs) for antitumor applications typically involves one protein domain binding to a tumor-associated antigen (TAA) (e.g., BCMA, CD19, DLL3, etc. [4]), while the other domain engages a target that promotes antitumor activity. Antitumor effects can be generally achieved through three primary mechanisms: (1) Direct binding to surface antigens of immune effector cells (e.g., CD3 on endogenous T cells, CD16 on NK cells, or CD47 on macrophages); (2) Engagement with receptors regulating T-cell responses; or (3) Interaction with other signaling pathways [5].
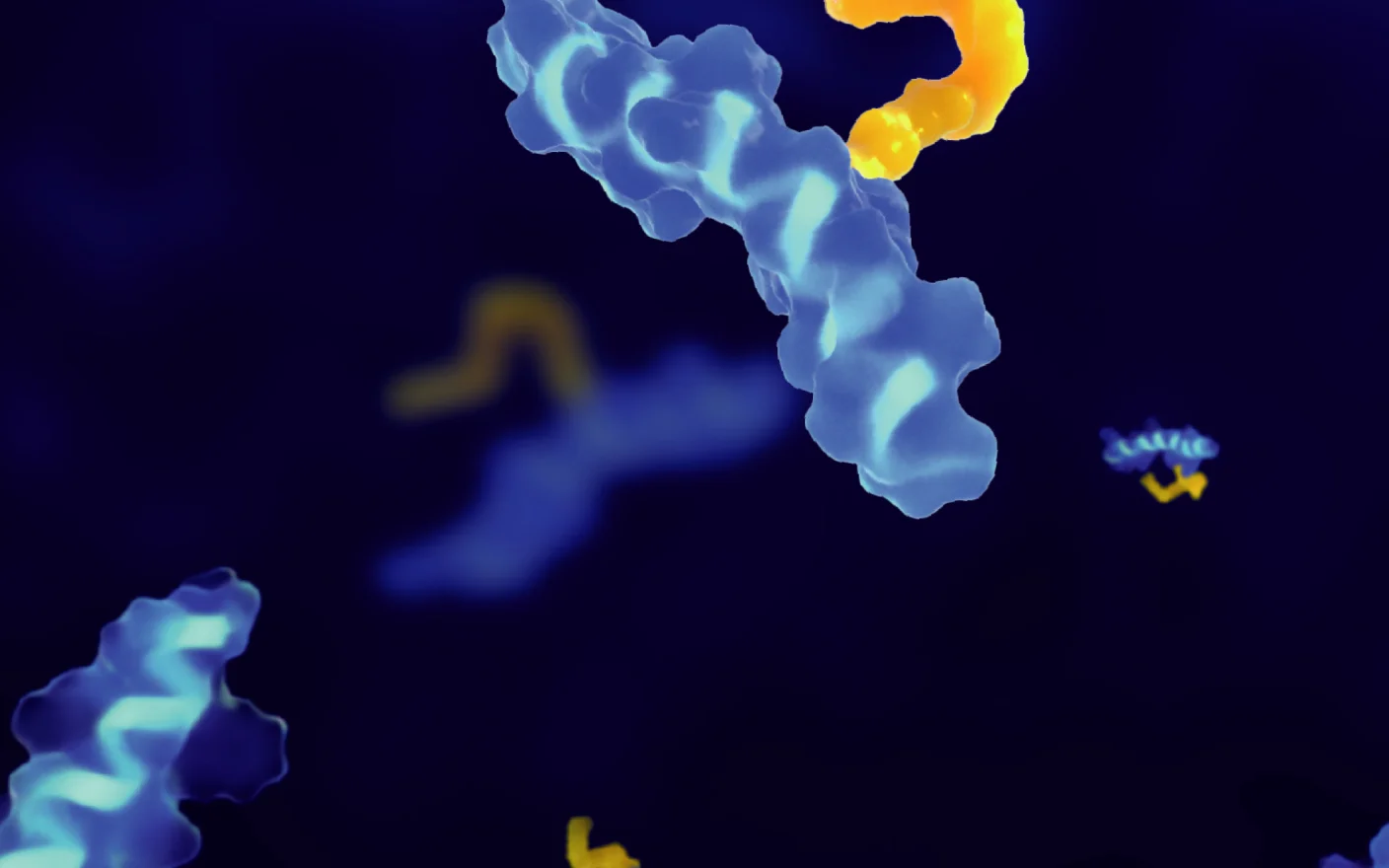
The most common bispecific T-cell engagers design employs the first approach, simultaneously targeting a TAA and a T-cell surface antigen. Due to the invariant CD3 chain of the T-cell receptor (TCR), CD3 is generally selected as the T-cell target. By bridging TAA and CD3, TCEs directly connect T cells to the cancer cells, enabling perforin and granzyme release independent of major histocompatibility complex (MHC) restriction and exerting potent tumor-killing effects.
Based on structural and mechanistic properties, bispecific T-cell engagers can be categorized into two major formats: those with or without an Fc domain. Fc-containing TCEs exhibit longer half-lives and greater stability. However, the intact Fc domain may impede T-cell migration and compromise antitumor activity [6]. Additionally, Fc-mediated antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) effects can trigger undesirable off-target immune reactions during systemic circulation.
In contrast, Fc-free TCE molecules have significantly smaller molecular sizes. Although displaying shorter half-lives in plasma, their enhanced diffusivity and tissue penetration may facilitate lower clinical dosing. Common examples of Fc-free TCEs include nanobodies and BiTE, composed of two single-chain variable fragments (scFvs) derived from anti-TAA and anti-CD3 monoclonal antibodies, linked via a short peptide spacer [1].

Figure 1. Structure and Mechanism of BiTE [7]
Indications and Advantages of TCEs
Compared to CAR-T therapy, TCEs offer a compact yet effective alternative. In addition to eliminating the need for genetic modification and ex vivo cell expansion required by CAR-T, TCEs demonstrate superior tumor cell killing efficiency as well. The key advantages of TCEs include 1) Serial tumor cell killing: a single T cell can serially engage multiple tumor cells via TCE bridging over time, enabling continuous tumor lysis. 2) T cell activation and expansion: TCE-mediated T cell activation triggers cytokine release and T cell proliferation, expanding the pool of T cells available for tumor killing. 3) MHC-independent recognition: by bypassing MHC restriction, TCEs could overcome a major immune evasion mechanism, which is tumor downregulation of MHC class I expression [3]. Therefore, due to their efficiency and flexibility, TCEs are treated as a pivotal research focus in contemporary cancer immunotherapy.
Regarding the recent advances of TCEs, their indication has expanded from hematologic malignancies targeting B-cell antigens (e.g., CD19, CD20) to solid tumors through novel TAA targeting. Moreover, their profound B cell-depleting capacity has spurred exploration in B cell-driven autoimmune diseases, including rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), highlighting their versatile therapeutic potential..
Clinical Applications of Approved TCE Therapeutics
Currently, most marked TCE drugs are indicated for hematologic malignancies. Their therapeutic targets include CD19, CD20, BCMA, and GPRC5D, with approved indications covering B-cell acute lymphoblastic leukemia (ALL), follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL), and multiple myeloma.
A key reason for TCEs’ predominant use in hematologic cancers lies in their manageable on-target/off-tumor toxicity profile. Several antigens like CD19/CD20 are also expressed on normal cells, and the resulting adverse effects are more clinically controllable in hematologic malignancies compared to solid tumors. For example, CD19-targeted therapy may cause B-cell aplasia and hypogammaglobulinemia, significantly increasing infection risk. However, such infections can be effectively controlled via clinical vigilance and the prompt use of antibiotics [5].
Table 1. FDA/EMA/NMPA Approved T-cell engagers [8].
Drug Name (Brand) | Target | Indication | Developer | Platform | Approval Date |
Catumaxomab (REMOVAB) | EpCAM×CD3 | Malignant ascites/pleural effusion | Neovii Biotech GmbH/Lingteng Medicine | Triomab | 2009.05 (EMA)* |
Blinatumomab ( Blincyto®) | CD19×CD3 | Precursor B-cell ALL, B-cell ALL | Amgen/BeiGene | BiTE | 2014.12 (FDA), 2015.11 (EMA), 2020.12 (NMPA) |
Tebentafusp (Kimmtrak®) | gp100×CD3 | Uveal melanoma | Immunocore | – | 2022.01 (FDA) |
Teclistamab (Tecvayli®) | BCMA×CD3 | Multiple myeloma | Genmab/Janssen | DuoBody | 2022.08 (EMA), 2022.10 (FDA), 2024.06 (NMPA) |
Mosunetuzumab (Lunsumio®) | CD20×CD3 | Follicular lymphoma | Roche | CrossMab/KIH | 2022.06 (EMA), 2022.12 (FDA), 2024.12 (NMPA) |
Epcoritamab (Epkinly®) | CD20×CD3 | DLBCL, HGBCL, PMBCL, FL | AbbVie/Genmab | DuoBody | 2023.05 (FDA) |
Glofitamab (Columvi®) | CD20×CD3 | DLBCL | Roche | CrossMab/KIH | 2023.06 (FDA), 2023.11 (NMPA) |
Talquetamab (Talvey®) | GPRC5D×CD3 | Multiple myeloma | Janssen | DuoBody | 2023.08 (FDA), 2025.02 (NMPA) |
Elranatamab (Elrexfio®) | BCMA×CD3 | Multiple myeloma | Pfizer | DuoBody | 2023.08 (FDA), 2025.03 (NMPA) |
Tarlatamab (Imdelltra®) | DLL3×CD3 | Small-cell lung cancer | Amgen | HLE BiTE | 2024.05 (FDA) |
Odronextamab (Ordspono™) | CD20×CD3 | Follicular lymphoma, DLBCL | Regeneron | Veloci-Bi | 2024.08 (EMA) |
*Withdrawn in 2017
Research Challenges and Advances in TCE Therapeutics
Apart from the above-mentioned reasons, the development of TCEs drugs targeting solid tumors faces multiple challenges, including cytokine release syndrome (CRS), complex tumor microenvironments (TME), limited T cell accessibility, as well as tumor antigen loss and lack of tumor antigen specificity [7, 9]. However, given that solid tumors constitute over 90% of all cancer types, it remains a key research area for TCE drugs.
Current advances in TCE therapies for solid tumors include, by are not limited to:
Reducing CRS by decreasing CD3 affinity and/or adjusting the number and affinity of TAAs [9];
Overcoming on-target off-tumor toxicity by targeting intracellular tumor neoantigens or designing conditionally activated TCE molecules [10, 11];
Enhancing anti-tumor activity while avoiding interference from antigen loss by increasing TAA targets (designing trispecific antibodies) [12];
Achieving TCE molecule accumulation at tumor sites via in situ mRNA expression [13];
Improving tumor-killing efficacy and preventing immune evasion through combination therapy with anti-PD-1/PD-L1 monoclonal antibodies or designing a PD-L1-targeting TCE molecule.
Disrupting immunosuppressive TME through combination therapy with adenosine receptor antagonists or IL-10 inhibitors [9].
Currently, multiple pharmaceutical companies are actively developing trispecific TCE therapeutics. For instance, Novartis has developed PIT565 (CD19×CD2×CD3), while GSK has also introduced the domestically developed trispecific molecule CMG1A46 (CD19×CD20×CD3). Recently, AstraZeneca's trispecific TCE molecule AZD5492 (CD20×TCR×CD8) for B-cell non-Hodgkin lymphoma and SLE has received multiple clinical approvals in China.
Pharmacokinetic (PK) Considerations for TCEs
PK Analysis
To date, regulatory agencies have not yet issued specific guidelines for TCE drugs. Therefore, pharmacokinetic studies of TCE drugs may refer to FDA’s draft guidance: Bispecific Antibody Development Programs (April 2019), and NMPA’s draft for comment: Technical Guidelines for Clinical Development of Bispecific Antibody Anti-Tumor Drugs (April 2022).
Regarding PK/ADA (anti-drug antibody) assay design and bioanalysis strategies for TCEs, refer to: Marketed Bispecific Antibodies and Their Pharmacokinetic Characteristics.
Cytokine Monitoring
Beyond standard PK/ADA assessment for bispecific antibodies, T-cell engagers often induce severe cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS), which normally co-occur. Thus, cytokine detection is particularly critical in preclinical studies of T-cell engagers [14]. Key cytokines include those from helper T cell subsets (Th1/Th2/Th17). Additional cytokines/chemokines may also be required depending on the drug targets. For example, the cytokine monitoring within blinatumomab (CD19×CD3) development includes IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, TNF-α, and IFN-γ, with IL-6, IL-10, and IFN-γ showing particularly marked elevation[15].
Due to the transient nature of cytokine release, dense sampling is advised at pre-dose, within hours post-first/last dose, and at 24h. Moreover, special attention should be given to mechanism-related cytokines.
WuXi AppTec DMPK offers cytokine profiling via flow cytometry-based CBA (cytometric bead array) and electrochemiluminescence-based MSD (Meso Scale Discovery), both enabling multiplexed detection with minimal sample volume. Notably, the MSD platform could provide detection sensitivity at the fg/mL level, addressing CBA’s limitations in sensitivity.
Immunophenotyping
To establish robust PK/PD models for TCE drugs, monitoring T-cell (and other major immune subsets) counts, T-cell activation/proliferation, and target cell numbers is essential. For example, both preclinical and clinical studies of blinatumomab revealed transient peripheral T-cell depletion due to endothelial adhesion or tissue migration. In addition, concurrent monitoring of B cells, NK cells, and monocytes is commonly conducted to assess drug-mediated cytolysis. These assays normally rely on flow cytometry-based immunophenotyping techniques.
WuXi AppTec DMPK has established robust immunophenotyping platforms for whole blood, bone marrow, and various tissues. For T-cell engagers, standardized flow cytometry panels have been developed to quantify T/B/NK cell populations and monocyte subsets, aiming to provide critical preclinical data support.
Case Study
As mentioned previously, designing conditionally activated T-cell engagers (antibody prodrug) to overcome on-target off-tumor toxicity represents one of the key directions in current TCE optimization strategies. In 2019, Seagen (now Pfizer) published an innovative universal antibody-masking approach in Nature Biotechnology [16] using high-affinity heterodimeric coiled-coil domains fused to the antibody N-terminus, enabling reversible inhibition of TCE’s biological activities. Upon exposure to tumor-associated proteases (e.g., MMP-2 and MMP-9), the coiled-coil peptides are cleaved, which selectively restores antibody activity while significantly reducing off-target toxicity risks.

Figure 2. Schematic diagram of antibody prodrug [17].
")
Figure 3. A. Binding of masked vs. unmasked anti-mouse CD3 antibody to CD3+ HT-2 cells. B. PK profiles of masked vs. unmasked anti-mouse CD3 antibody (0.5 mg/kg IV in BALB/c mice)[16].
Given the known CD3-targeting toxicity, researchers validated whether the coiled-coil masking peptide could effectively block antibody binding to CD3 proteins on lymphocyte surfaces. As shown in Figure 3A, at concentrations up to 2 μM, the masking peptide effectively prevented the anti-mouse CD3 antibody from binding to CD3+ HT-2 cells.
Furthermore, after intravenous administration of 0.5 mg/kg anti-mouse CD3 antibody to BALB/c mice (n=3), the unmasked antibody became undetectable in plasma within 2 days due to the antigen sink effect caused by widespread CD3 expression. In contrast, the masked anti-mouse CD3 antibody exhibited excellent circulation stability over 15 days (Figure 3B).

Figure 4. Dynamic concentrations of IL-2 and IFN-γ in mice post-dosing masked or unmasked anti-mouse CD3 antibodies[16].
Simultaneously, the masking strategy effectively mitigated the excessive release of proinflammatory cytokines. As shown in Figure 4, the unmasked parental antibody significantly elevated IL-2 and IFN-γ levels in animals within 4 hours post-dose. In contrast, the masked anti-mouse CD3 antibody substantially reduced this on-target off-tumor toxicity.
This helical peptide-based masking strategy offers universal applicability, high-efficiency masking, tumor-selective activation, and optimized PK characteristics, expanding the therapeutic window of T-cell engagers and enabling clinical dose escalation. It unlocks those targets previously deemed undruggable, and potentially accelerates the development and clinical translation of T-cell engagers.
The Bottom Line
T-cell engagers have emerged as a promising modality in cancer therapy for their ability to potently activate T cells and precisely eliminate tumor cells. While multiple bispecific T-cell engagers have demonstrated efficacy in hematologic malignancies, advances in molecular engineering, multi-targeting strategies, and combination therapies (e.g., with checkpoint inhibitors) are unlocking their potential in solid tumors. With accumulating clinical data and ongoing technological innovation, TCE therapeutics are expected to benefit a broader population of cancer patients.
WuXi AppTec DMPK offers integrated bioanalytical platforms for T-cell engagers, including PK, ADA, cytokine profiling, immunophenotyping, and clinicopathological testing, providing end-to-end support for TCE drug discovery and development.
Authors: Xiaoqi Wang, Huan Yan, Jie Pan, Qigan Cheng, Jing Jin
Talk to a WuXi AppTec expert today to get the support you need to achieve your drug development goals.
Committed to accelerating drug discovery and development, we offer a full range of discovery screening, preclinical development, clinical drug metabolism, and pharmacokinetic (DMPK) platforms and services. With research facilities in the United States (New Jersey) and China (Shanghai, Suzhou, Nanjing, and Nantong), 1,000+ scientists, and over fifteen years of experience in Investigational New Drug (IND) application, our DMPK team at WuXi AppTec are serving 1,600+ global clients, and have successfully supported 1,700+ IND applications.









Related Services and Platforms




Stay Connected
Keep up with the latest news and insights.

