

2024 FDA-Approved Drug Highlights: Overview of Human Pharmacokinetics and DDI Profiles
-

Articles
-

Apr 29, 2025
In 2024, the U.S. Food and Drug Administration (FDA) approved 50 new small-molecule and biologic drugs, spanning diverse therapeutic areas and molecular classes[1]. This article analyzes 12 representative drugs, focusing on their drug metabolism and pharmacokinetics (DMPK) and drug interaction (DDI) characteristics. Highlighted therapies include Rezdiffra (resmetirom, the first drug approved for non-alcoholic steatohepatitis [NASH]/metabolic dysfunction-associated steatohepatitis [MASH]), Lazcluze (lazertinib, for EGFR-mutant non-small cell lung cancer), Cobenfy (xanomeline/trospium chloride, for schizophrenia), Alyftrek (vanzacaftor/tezacaftor/deutivacaftor, for cystic fibrosis), and Winrevair (sotatercept-csrk, for pulmonary arterial hypertension). This review provides pharmacokinetic data and underscores clinical implications across molecular types and indications.
Overview of 2024 FDA-approved drugs
The FDA’s Center for Drug Evaluation and Research (CDER) approved 50 new drugs in 2024 (Click to access the full detailed list: Summary of Human ADME Data for 50 FDA-Approved Drugs in 2024.), slightly fewer than 2023’s 55 approvals but exceeding the 10-year rolling average of 46.5.
Key statistics:
Priority review: 56%
Orphan drug designation: 52%
Breakthrough therapy designation: 36%
Accelerated approval (surrogate endpoints): 14%
1. Molecular diversity (Figure 1):
Small molecules: 60% (30 drugs), predominantly oncology agents (23%).
Biologics: 32% (16 drugs), including 13 monoclonal/bispecific antibodies (e.g., tislelizumab-jsgr from BeiGene).
TIDES: 4 drugs (2 oligonucleotides, 2 peptides).
PEGylated/deuterated drugs: 4 drugs (2 deuterated small molecules, 2 PEGylated peptides).
Fluorinated/N-aromatic heterocycles: 66% of small molecules.
2. Therapeutic areas (Table 1, Figure 2):
Oncology: 14 drugs (7 small molecules, 6 antibodies, 1 cytokine fusion protein).
Cardiovascular: 4 drugs (3 small molecules, 1 antisense oligonucleotide).
Central nervous system: 2 drugs.
Rare diseases: e.g., Alyftrek (cystic fibrosis), Tryngolza (familial chylomicronemia syndrome).

Figure 1. Number of FDA-approved drugs in 2024 by molecular type
Table 1. Number of FDA-approved drugs in 2024 by disease area
Disease areas | Total | Small molecules | Antibodies | Others |
Oncology | 14 | 7 | 6 | 1 (cytokine fusion protein) |
Cardiovascular | 4 | 3 | 0 | 1 (antisense oligonucleotide) |
Central nervous system (CNS) | 2 | 1 | 1 | 0 |
Dermatology | 5 | 2 | 2 | 1 (protein toxin) |
Hematology | 5 | 0 | 4 | 1 (oligonucleotide) |
Nephrology | 1 | 1 | 0 | 0 |
Hepatology | 3 | 3 | 0 | 0 |
Respiratory | 2 | 2 | 0 | 0 |
Endocrinology | 3 | 2 | 0 | 1 (peptide) |
Infectious diseases | 3 | 3 | 0 | 0 |
Other | 8 | 6 | 0 | 2 |

Figure 2. FDA-Approved drugs by molecular type and disease area
Overall, the FDA’s drug approvals in 2024 highlight the importance of biologics, TIDES drugs, fluorinated compounds, and N-heterocyclic compounds in drug development, as well as the ongoing development of combination therapies and imaging diagnostic drugs.
Detailed pharmacokinetic and DDI profiles of 12 FDA-approved drugs
Lazcluze™ (lazertinib)
Indication: Non-smallcell lung cancer (combined with amivantamab).
Mechanism: Epidermalgrowth factor receptor (EGFR) kinase inhibitor.
Molecular type: Small-molecule.
Approval date: August 19, 2024.
Company: Johnson & Johnson.
Route: Oral (tablet).

Pharmacokinetics & drug interactions:
Absorption:
Dose-proportional increases in Cmax and AUC from 20 mg to 320 mg (0.08–1.3x the approved dose).
Steady-state plasma exposure achieved by Day 15 (AUC accumulation ratio: 2x).
Tmax: 2–4 hours.
Distribution:
Volume of distribution (Vd): 2680 L (51% CV).
Plasma protein binding: 99.2%.
Metabolism:
Half-life (t½): 3.7 days (56% CV).
Clearance (CL): 36.4 L/h (47% CV).
Primarily metabolized via glutathione conjugation and CYP3A4.
Excretion:
Feces: 86% (≤5% unchanged).
Urine: 4% (≤0.2% unchanged).
Drug interactions:
Inhibits CYP3A4, UGT1A1, BCRP, and OCT1 in vitro.
Strong CYP3A4 inducers (e.g., rifampin): ↓Cmax by 72%, ↓AUC by 83%.
Strong CYP3A4 inhibitors (e.g., itraconazole): ↑Cmax by 1.2x, ↑AUC by 1.5x.
CYP3A4 substrates (e.g., midazolam): ↑Midazolam Cmax by 1.4x, AUC by 1.5x.
BCRP substrates (e.g., rosuvastatin): ↑Rosuvastatin Cmax by 2.2x, AUC by 2x.
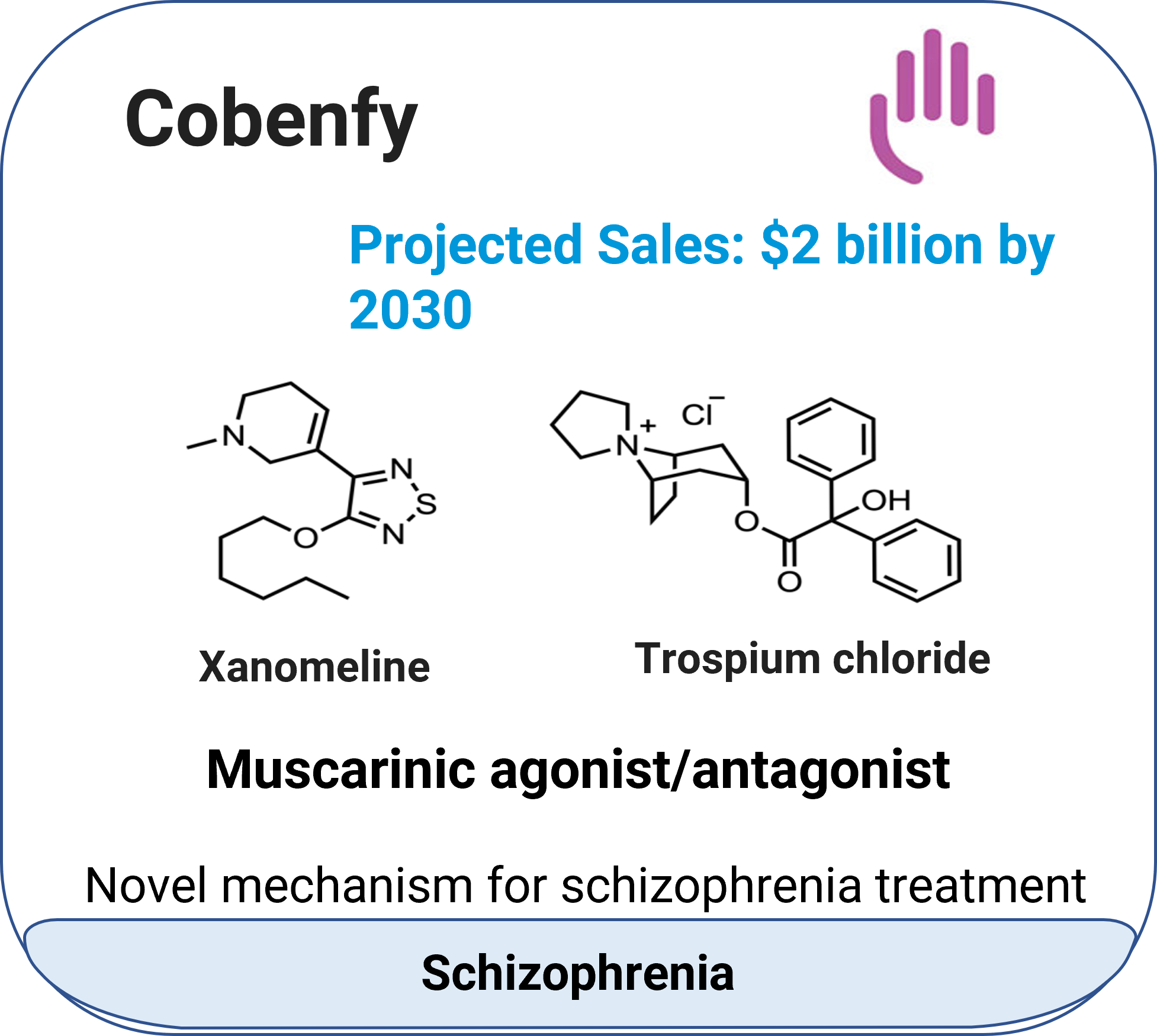
Cobenfy™ (xanomeline + trospium chloride)
Indication: Schizophrenia
Mechanism:
Xanomeline: Central M1/M4 muscarinic agonist;
Trospium chloride: Peripheral muscarinic antagonist.
Molecular type: Small-molecule combination.
Approval date: September 26, 2024.
Company: Bristol Myers Squibb.
Route: Oral (capsule).
Pharmacokinetics & drug interactions:
Absorption:
Xanomeline:
Tmax: 2 hours.
High-fat meal ↑AUC by 30% (no change in Cmax).
Trospium chloride:
Tmax: 1 hour.
High/low-fat meals ↓AUC by 85–90%, ↓Cmax by 70–75%.
Distribution:
Xanomeline: Vd = 10,800 L; plasma protein binding = 95%.
Trospium chloride: Vd = 531 L; plasma protein binding = 80%.
Metabolism:
Xanomeline:
t½: 5 hours.
CL: 1950 L/h.
Metabolized by CYP2D6, CYP2B6, CYP1A2, CYP2C9/19, and FMO1/3.
Trospium chloride:
t½: 6 hours.
CL: 796 L/h.
Metabolized via ester hydrolysis and glucuronidation.
Excretion:
Xanomeline: Urine (78%, <0.01% unchanged); feces (12%).
Trospium chloride: Urine (85–90% unchanged).
Drug interactions:
Risk of increased adverse effects with CYP2D6 inhibitors, sensitive substrates of CYP3A4 or P-gp, or drugs eliminated via active renal tubular secretion.
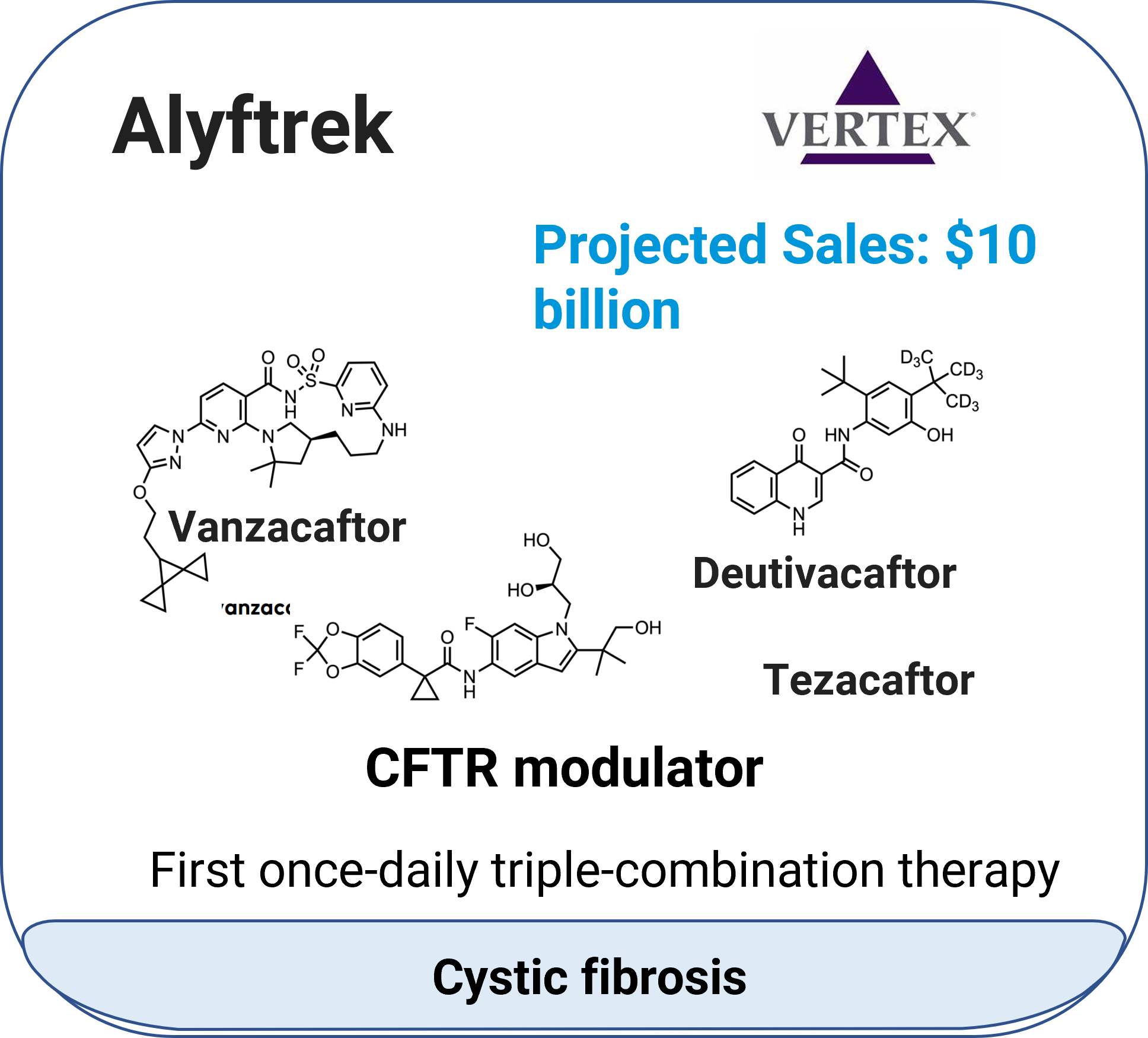
Alyftrek™ (vanzacaftor + tezacaftor + deutivacaftor)
Indication: Cystic fibrosis.
Mechanism: CFTR modulators.
Molecular type: Small-molecule combination.
Approval date: December20, 2024.
Company: Vertex Pharmaceuticals.
Route: Oral (tablet).
Pharmacokinetics & drug interactions:
Absorption (12+ years):
Vanzacaftor:
Cmax,ss: 0.812 μg/mL; AUC0-24h,ss: 18.6 μg·h/mL.
Tmax: 7.8 hours (3.7–11.9 hours).
Food effect: ↑AUC by 4x (low-fat meals), 6x (high-fat meals).
Tezacaftor:
Cmax,ss: 6.77 μg/mL; AUC0-24h,ss: 89.5 μg·h/mL.
Tmax: 1.6 hours (1.4–1.7 hours).
Deutivacaftor:
Cmax,ss: 2.33 μg/mL; AUC0-24h,ss: 39.0 μg·h/mL.
Tmax: 3.7 hours (2.7–11.4 hours).
Food effect: ↑AUC by 3x (low-fat meals), 4x (high-fat meals).
Distribution:
Vanzacaftor: Vd = 121 L; plasma protein binding >99%.
Tezacaftor: Vd = 73.1 L; plasma protein binding ~99%.
Deutivacaftor: Vd = 159 L; plasma protein binding >99%.
Metabolism:
All three components were metabolized by CYP3A4/5.
Vanzacaftor: t½ = 92.8 hours; CL = 1.34 L/h.
Tezacaftor: t½ = 22.5 hours; CL = 1.22 L/h.
Deutivacaftor: t½ = 19.2 hours; CL = 7.29 L/h.
Excretion:
Vanzacaftor: Feces (91.6%, metabolites); urine (0.5%).
Tezacaftor: Feces (72%); urine (13.7%).
Drug interactions:
In vitro, vanzacaftor is a substrate of CYP3A, and inhibits BCRP and P-gp; Tezacaftor is a substrate of CYP3A, P-gp, BCRP and OATP1B1, and inhibits P-gp; Deutivacaftor is a substrate of CYP3A and P-gp, and inhibits CYP2C8/2C9/3A4, P-gp and BCRP.
Avoid strong/moderate CYP3A4 inducers (↓efficacy) or inhibitors (↑toxicity).
Rezdiffra™ (resmetirom)
Indication: Noncirrhotic non-alcoholic steatohepatitis with moderate to advanced liver scarring.
Mechanism: Thyroid hormone receptor-β (THR-β) agonist.
Molecular type: Small molecule.
Approval date: March 14, 2024.
Company: Madrigal Pharmaceuticals.
Route: Oral (tablet).

Pharmacokinetics & drug interactions:
Absorption:
Steady state achieved in 3–6 days.
80 mg/day: Cmax,ss = 778 ng/mL (41.5% CV); AUCtau,ss = 5850 ng·h/mL (60.5% CV).
100 mg/day: Cmax,ss = 971 ng/mL (40.9% CV); AUCtau,ss = 7780 ng·h/mL (65.5% CV).
Food reduces Cmax by 33% and delays Tmax by 2 hours.
Distribution: Vd = 68 L (227% CV); plasma protein binding >99%.
Metabolism:
t½ = 4.5 hours; CL = 17.5 L/h (56.3% CV).
Metabolized by CYP2C8.
Excretion: Feces (67%); urine (24%).
Drug interactions:
Inhibits CYP2C8, UGT1A4/1A9, OATP1B1/1B3, BCRP, OAT3, and BSEP in vitro.
A substrate of OATP1B1/1B3 and BCRP in vitro.
Clopidogrel (CYP2C8 inhibitor): ↑Resmetirom AUC by 1.7x.
Piogkitazone (CYP2C8 substrate): ↑Piogkitazone AUC by 1.5x.
Statins (e.g., simvastatin): ↑Simvastatin AUC by 1.7x.
Voranigo™ (vorasidenib)
Indication: IDH-mutant grade 2 astrocytoma or oligodendroglioma.
Mechanism: :IDH1/2 inhibitor.
Molecular type: Small molecule.
Approval date: August 6, 2024.
Company: Servier Pharmaceuticals.
Route: Oral (tablet).

Pharmacokinetics & drug interactions:
Absorption:
Dose-proportional exposure (10–200 mg).
At the highest approved recommended dosage (50 mg/day): Cmax= 133 ng/mL (73% CV); AUC = 1988 h·ng/mL (95% CV).
Tmax: 2 hours (0.5–4 hours).
Absolute bioavailability: 34%.
Food effect: High-fat and high-calorie meal ↑Cmax by 3.1x, AUC by 1.4x.
Low-fat and low-calorie meal ↑Cmax by 2.3x, AUC by 1.4x
Distribution:
Vd = 3930 L (40% CV); human plasma protein binding = 97%.
Brain tumor-to-plasma ratio: 1.6.
Metabolism:
t½ = 10 days (57% CV); CL = 14 L/h (56% CV).
Primarily metabolized by CYP1A2; non-CYP pathways account for 30%.
Excretion: Feces: 85% (56% unchanged); urine: 4.5%.
Drug interactions:
Vorasidenib is an inducer of CYP2B6, CYP2C8, CYP2C9, CYP2C19 and CYP3A and UGT1A4.
Vorasidenib is an inhibitor of BCRP.
Concomitant use with moderate CYP1A2 inhibitors (e.g., ciprofloxacin): ↑ Cmax 1.3x, ↑AUC 2.5x.
Concomitant use with strong CYP1A2 inhibitors (e.g., fluvoxamine): ↑AUC ≥5x.
Concomitant use with moderate CYP1A2 inducers (e.g., phenytoin or rifampicin): ↓Cmax 30%, ↓AUC 40%.
Tryvio™ (aprocitentan)
Indication: Hypertension
Mechanism: Dual endothelin (ETA/ETB) receptor antagonist.
Molecular type: Small molecule.
Approval date: March 19, 2024.
Company: Idorsia Pharmaceuticals.
Route: Oral (tablet).

Pharmacokinetics & drug interactions:
Absorption:
Single 25 mg dose: Cmax = 1.3 μg/mL (19% CV), Tmax = 4–5 hours, AUC0-tau=23 μg·h/mL (17% CV);
Steady state achieved by Day 8 (accumulation ratio: 3x).
Distribution:
Vd = 20 L; plasma protein binding >99%.
Whole blood-to-plasma ratio: 0.63.
Metabolism:
t½ = 41 hours; CL = 0.3 L/h.
Metabolized by UGT1A1- and UGT2B7-mediated N-glucosidation and non-enzymatic hydrolysis
Excretion: Feces (25%, 6.8% unchanged); urine (52%, 0.2% unchanged).
Drug interactions:
Inhibits CYP3A4, CYP2C family, BCRP, BSEP, and NTCP.
Inducer of CYP3A4
Substrate and inhibitor of UGT1A1 and UGT2B7.
Substrate of P-gp and BCRP
Concomitant administration with UGT inducers: ↓aprocitentan exposure.
Rytelo™ (imetelstat)
Indication: Low-to intermediate-1 risk myelodysplastic syndromes.
Mechanism: An oligonucleotide human telomerase inhibitor that binds to the template region ofthe RNA component of human telomerase (hTR), inhibits telomerase enzymatic activity, and prevents telomere binding.
Molecular type: Oligonucleotide.
Approval date: June 6, 2024.
Company: Geron Corporation.
Route: Intravenous (infusion).

Pharmacokinetics & drug interactions:
Absorption:
7.1 mg/kg dose every 4 weeks: Cmax = 18.3 μM (27.3% CV); AUC0-28d = 114.2 h·μM (43.2% CV).
No accumulation after 4-week cycles.
Distribution:
Vd = 14.1 L (27.2% CV); human plasma protein binding >94%.
Metabolism:
t½ = 4.9 hours (43.2% CV).
Metabolized by nucleases to nucleotides of various lengths.
Excretion: Data not provided.
Drug interactions:
Inhibits OATP1B1 and OATP1B3; not an inhibitor or inducer of CYP450 enzymes.
Tryngolza™ (olezarsen)
Indication: Familial chylomicronemia syndrome.
Mechanism: An ASO-GalNAc3 conjugate that binds to apoC-III mRNA, leading to mRNA degradation and resulting in a reduction of serum apoC-III protein. Reduction of apoC-IIIprotein leads to increased clearance of plasma TG and VLDL.
Molecular type: Oligonucleotide (GalNAc3-conjugated).
Approval date: December 19, 2024.
Company: Ionis Pharmaceuticals.
Route: Subcutaneous (injection).

Pharmacokinetics & drug interactions:
Absorption:
80 mg/month: Cmax = 883 ng/mL (SD 662); AUCτ = 7440 ng·h/mL (SD 3880).
Tmax: 2 hours; no accumulation.
Distribution:
Vd (central) = 91.9 L; Vd (peripheral) = 2960 L.
plasma protein binding>99%
Primarily distributed to the liver and kidneys.
Metabolism:
t½: ~4 weeks.
metabolized by endo- and exonucleases to short oligonucleotide fragments of varying sizes within the liver.
Excretion: Urine (<1% unchanged within 24 hours).
Drug interactions:
No interactions with CYP450, transporters, or plasma proteins.
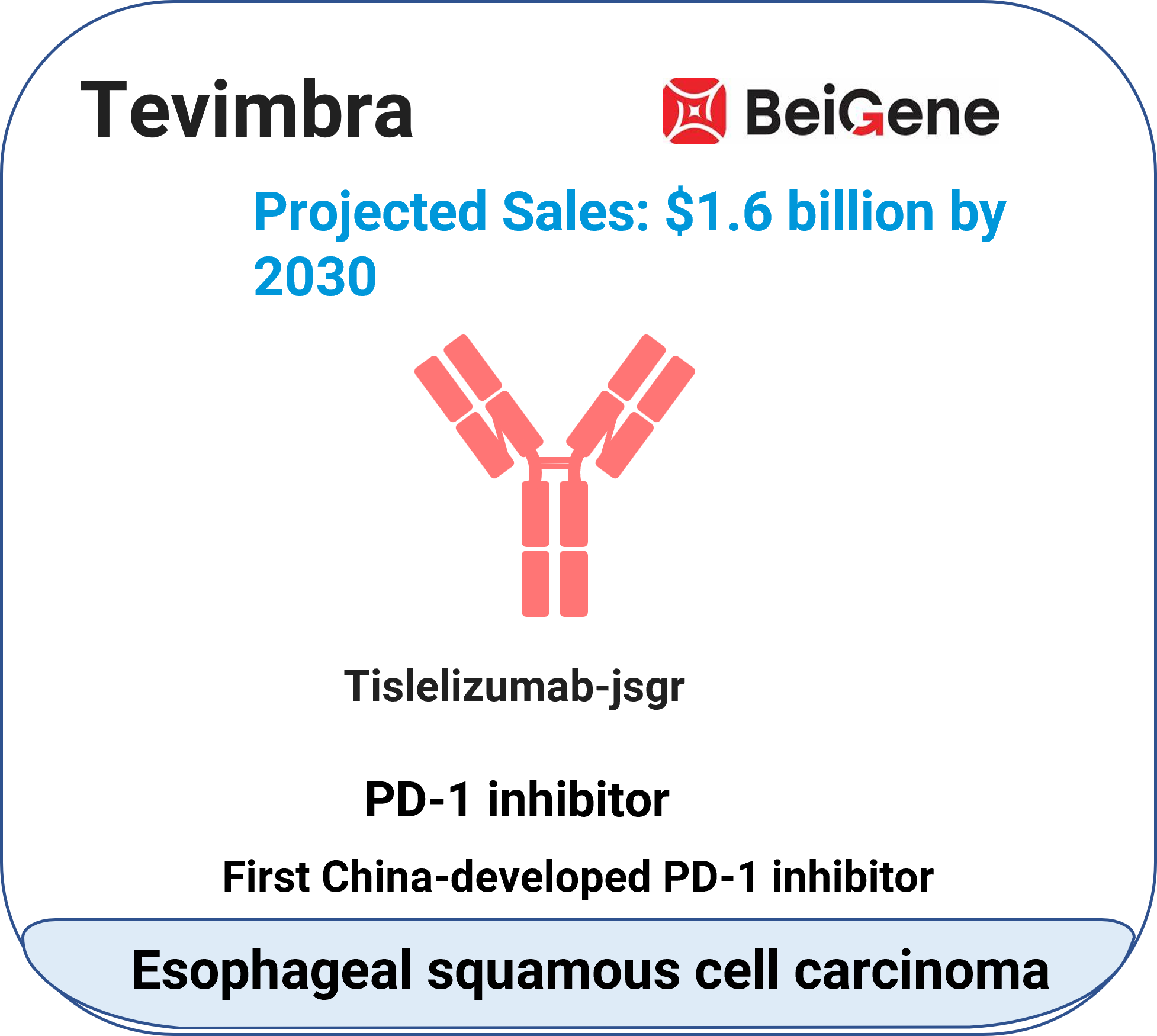
Tevimbra™ (tislelizumab-jsgr)
Indication: Unresectable/metastatic esophageal squamous cellcarcinoma.
Mechanism: PD-1 inhibitor (monoclonal antibody).
Molecular type: Monoclonal antibody.
Approval date: March 13, 2024.
Company: BeiGene.
Route: Intravenous (injection).
Pharmacokinetics:
Absorption:
200 mg every 3 weeks: Cmax = 110 μg/mL (22.2% CV); AUCtau = 1283 μg·d/mL (28.7% CV).
Steady state achieved by Week 12 (accumulation ratio: 2.14x).
Distribution: Vd = 6.42 L (32.6% CV).
Metabolism:
t½ = 24 days (31% CV); CL = 0.153 L/day (29.5% CV).
Degraded via proteolysis (similar to endogenous IgG).
Excretion: Data not provided.
Drug interactions: Data not provided.
Kisunla™ (donanemab-azbt)
Indication: Alzheimer’s disease.
Mechanism: Amyloid-beta-targeting monoclonal antibody.
Molecular type: Monoclonal antibody.
Approval date: July 2, 2024.
Company: Eli Lilly.
Route: Intravenous (injection).

Pharmacokinetics:
Absorption:
Dose: 700 mg → 1400 mg every 4 weeks.
Steady state reached after first dose (accumulation <1.3x).
Dose-proportional exposure (10–40 mg/kg).
Distribution: Vd (central) = 3.36 L.
Metabolism:
t½ = 12.1 days; CL = 0.0255 L/h.
Degraded via proteolysis.
Excretion: Negligible renal elimination.
Drug interactions: Data not provided.
Imdelltra™ (tarlatamab-dlle)
Indication: Extensive-stagesmall cell lung cancer.
Mechanism: DLL3/CD3 bispecific T-cell engager.
Molecular type: Bispecific antibody.
Approval date: May 16, 2024.
Company: Amgen.
Route: Intravenous (injection).

Pharmacokinetics & drug interactions:
Absorption:
Dose-proportional exposure (1–100 mg every 2 weeks).
Steady state (Cycle 2 Day 15): Cavg = 1040 ng/mL (44% CV); Cmax = 3400 ng/mL (40% CV).
Distribution: Vd = 8.6 L (18.3% CV).
Metabolism:
t½ = 11.2 days (4.3–26.5 days); CL = 0.65 L/day (44% CV).
Degraded into small peptides.
Excretion: Data not provided.
Drug interactions:
May transiently inhibit CYP450 enzymes due to cytokine release.
Winrevair™ (sotatercept-csrk)
Indication: Pulmon aryarterial hypertension.
Mechanism: Activin signaling inhibitor (recombinant fusion protein).
Molecular type: :Recombinant fusion protein.
Approval date: March 26, 2024.
Company: Acceleron Pharma & Merck.
Route: Subcutaneous (injection).

*The projected sales in the images are sourced from reference 6.
Pharmacokinetics:
Absorption:
0.7 mg/kg every 3 weeks: AUC = 172 μg·d/mL (34.2% CV); Cmax = 9.7 μg/mL (30% CV).
Steady state achieved at ~15 weeks (accumulation ratio: 2.2x).
Absolute bioavailability: 66%.
Tmax: 7 days.
Distribution: Vd = 5.3 L (27.3% CV); weight-dependent.
Metabolism:
t½ = 24 days; CL = 0.18 L/day (weight-dependent).
Degraded into small peptides.
Excretion: Data not provided.
Drug interactions: Data not provided.
Concluding remarks
By analyzing the pharmacokinetic and DDI characteristics of representative new drugs approved by the FDA in 2024, it can be observed that drugs of different molecular types and indications exhibit distinct advantages in ADME studies. The significant optimization of these ADME and DDI properties provides valuable insights and references for future drug development.
WuXi AppTec DMPK leverages its extensive expertise across small molecules, antibodies, nucleic acids, and peptides, with specialization in ADME screening and optimization, accelerating client projects into clinical stages while proactively identifying and mitigating potential clinical risks.
Authors: Yan Pan, Yunyi Yan, Ting Peng, Yu Wang, Lijuan Hou, Jing Jin
Talk to a WuXi AppTec expert today to get the support you need to achieve your drug development goals.
Committed to accelerating drug discovery and development, we offer a full range of discovery screening, preclinical development, clinical drug metabolism, and pharmacokinetic (DMPK) platforms and services. With research facilities in the United States (New Jersey) and China (Shanghai, Suzhou, Nanjing, and Nantong), 1,000+ scientists, and over fifteen years of experience in Investigational New Drug (IND) application, our DMPK team at WuXi AppTec are serving 1,600+ global clients, and have successfully supported 1,700+ IND applications.
Reference
[1] FDA.Novel Drug Approvals for 2024. http://www.fda.gov/drugs/novel-drug-approvals-fda/novel-drug-approvals-2024
[2] Mullard, A. 2024 FDA approvals. Nat Rev Drug Discov 24, 75–82 (2025).
[3] de la Torre, B. G. & Albericio, F. The Pharmaceutical Industry in 2024: An Analysis of FDA Drug Approvals from the Perspective of Molecules. Preprint at http://doi.org/10.20944/preprints202501.0253.v1 (2025).
[4] FDA & CDER. HIGHLIGHTS OF PRESCRIBING INFORMATION. www.fda.gov/medwatch.
[5] FDA. CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 217785Orig1s000 INTEGRATED REVIEW.
[6] 2024 drug approvals: Small companies loom large with several key FDA nods. http://www.fiercepharma.com/pharma/2024-drug-approvals










Related Services and Platforms




Stay Connected
Keep up with the latest news and insights.

